关注一种不可忽视的儿童肢体疼痛
时间:2025-08-09 12:27:49 热度:37.1℃ 作者:网络
论坛导读:Fabry病作为一种X连锁溶酶体贮积症,因GLA基因突变导致α-半乳糖苷酶A活性缺失,引起糖鞘脂代谢产物globotriaosylceramide(GL-3) 及其脱酰基形式lyso-GL3在多器官蓄积。Fabry危象是其最具破坏性的急性临床表现,以剧烈肢端疼痛为核心,伴随自主神经功能紊乱和多系统急性功能障碍。由于该病平均诊断延迟长达14年,危象常被误诊为“生长痛”或“功能性胃肠病”。
Fabry病是一种罕见的X连锁遗传性溶酶体贮积症,因GLA基因突变导致α-半乳糖苷酶A(α-Gal A)活性缺乏,引起鞘糖脂(如Gb3和Lyso-Gb3)在全身多器官沉积,造成进行性损伤。Fabry危象的本质是糖鞘脂神经毒性的急性加剧:①lyso-GL3直接损伤:作为神经毒性鞘脂,lyso-GL3在背根神经节和三叉神经节蓄积,通过激活TRPV1通道介导痛觉敏化,同时破坏线粒体能量代谢,诱发轴突变性。②微血管功能障碍:内皮细胞GL-3沉积导致微循环缺血,神经内膜血流减少引发缺氧性疼痛。③继发性炎症级联:沉积物质激活Toll样受体,TNF-α、IL-6等促炎因子释放,放大神经损伤。
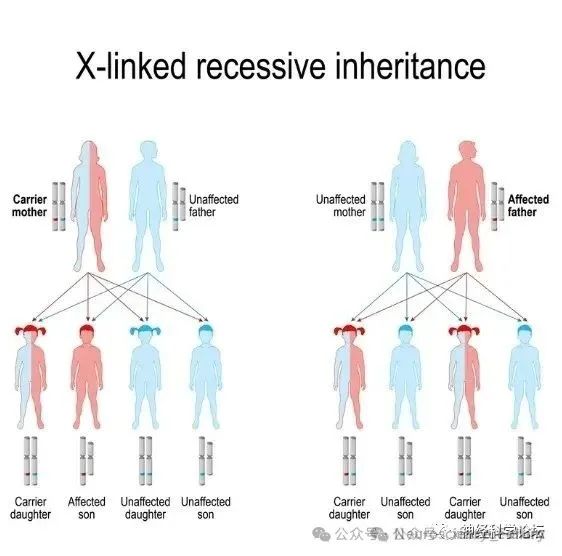
💡疾病定义与遗传机制
Fabry病遗传特性属于X连锁隐性遗传,GLA基因位于X染色体(Xq22.1),男性发病率(1/40,000~1/110,000)高于女性,男性多表现为典型症状,女性因X染色体随机失活呈症状轻重不一。遗传规律患病男性必然遗传给女儿(女儿为携带者),但不会遗传给儿子;女性携带者有50%概率遗传给子女。
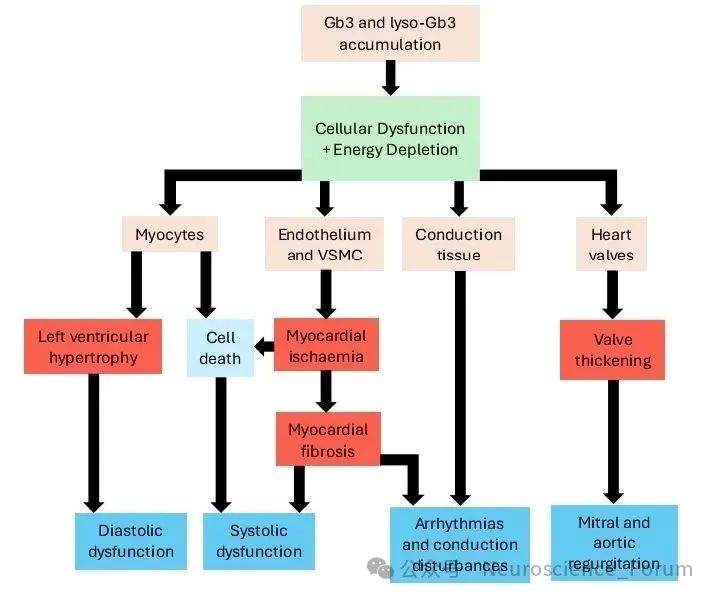
α-Gal A酶活性缺失→鞘糖脂在血管内皮、肾脏、心脏及神经细胞沉积→引发慢性炎症反应、纤维化及器官功能衰竭。未治疗者预期寿命缩短(男性减15-20年,女性减6-10年)。
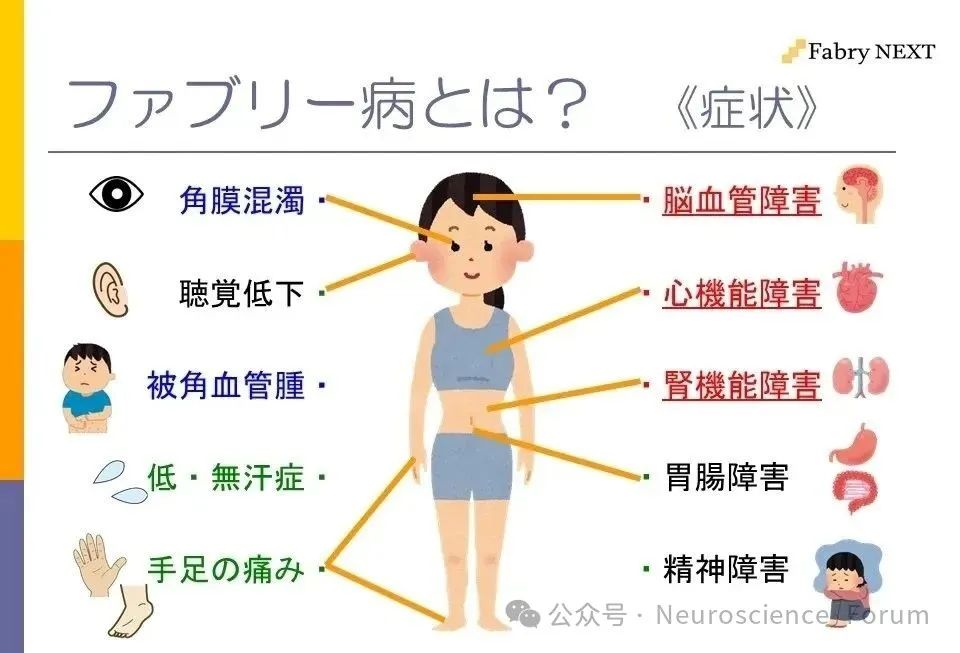
💡临床表现
多系统进行性损害早期症状(儿童/青少年期):①神经性疼痛:手足烧灼样剧痛(Fabry危象),常由发热或运动诱发。②皮肤病变:脐周、腹股沟区紫红色血管角质瘤。③其他:少汗/无汗、角膜涡状混浊、胃肠道症状(腹泻、腹痛)。
晚期(成年后):肾脏:蛋白尿→慢性肾衰竭(终末期肾病占比30%)。心脏:左心室肥厚(50%患者)、心律失常、心力衰竭。神经系统:脑白质病变、卒中样发作(青年卒中重要病因)。
表型异质性:经典型男性为主,儿童期起病,多系统受累;迟发型成年发病,以心脏或肾脏单一损害为主。
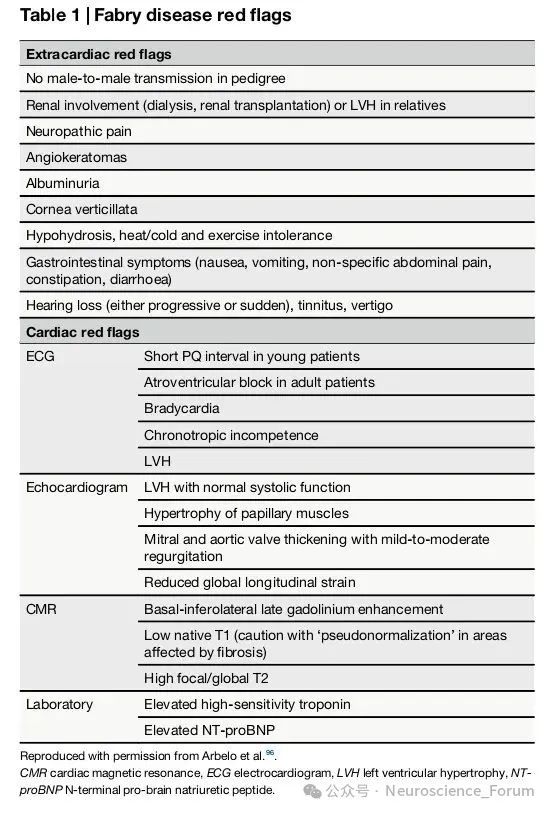
💡Fabry危象表现为多系统急性功能障碍
①疼痛特征:90%患者儿童期即出现,多由感染、应激、温度变化诱发,四肢烧灼痛呈“手套-袜套”分布,持续数小时至数日。
②自主神经危象:胃肠症状见于70% 患者,急性期表现为假性肠梗阻;心脏自主神经病变可致体位性低血压及晕厥。
③不典型表现:迟发型患者可首发心律失常危象(如Afib风暴)或肾性危象(急性蛋白尿激增)。
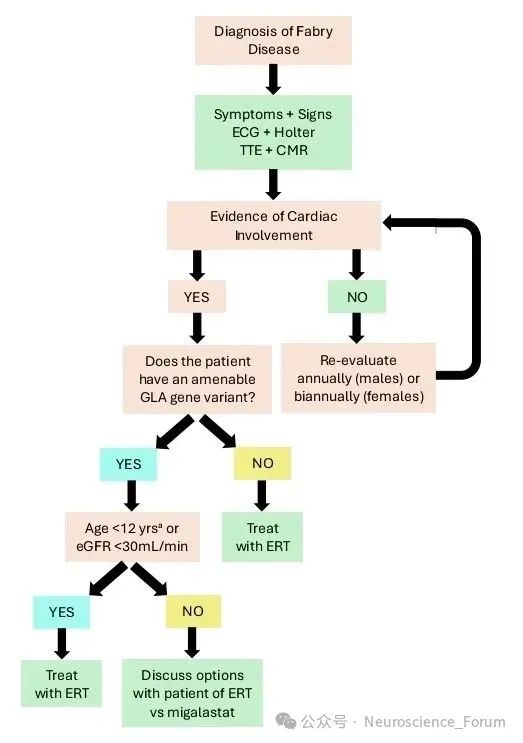
💡诊断方法
①酶活性检测:男性血浆/白细胞α-Gal A活性<5%正常值(筛查首选)。女性酶活性可能接近正常,需结合基因检测。
②生物标志物与基因检测:血浆Lyso-GL-3敏感度达98%,尤其适用于女性诊断。基因测序GLA基因突变分析为金标准(已发现超1000种突变)。 新技术三代测序(CAFD法)可检出传统方法遗漏的内含子变异,诊断率提升至97.9%。
③病理检查:组织活检(如肾脏、皮肤)可见嗜锇性包涵体(鞘脂沉积特征)。
💊 治疗策略
①特异性治疗:酶替代疗法(ERT)*重组α-Gal A(阿加糖酶α/β),每2周静脉输注1次,可延缓器官损伤,降低心肾事件风险。 建议30-40岁前启动治疗,逆转早期病变。
②分子伴侣疗法:口服药物(如米加司他)适用于特定错义突变(约35%患者)。
③基因治疗(突破性进展):rAAV基因药物ZS805注射液单次输注、动物研究可长期生效,尚未上市。
④对症支持治疗:神经痛卡马西平、加巴喷丁。
蛋白尿ACEI/ARB类药物。终末期肾病透析或肾移植。
⑤预后:规范治疗下中位生存期>50岁,但延迟诊断(平均13.5年)及经济负担(65%患者治疗受限)仍是主要挑战。
综上所述,Fabry危象的早期识别依赖于对隐匿性疼痛、自主神经及微血管功能障碍的警觉,结合多模态生物标志物与影像技术实现超前干预。作为罕见遗传病,早期诊断(新生儿筛查、家族基因检测)及ERT治疗是关键。未来需推进高危人群筛查(如心肾疾病早发群体)、开发神经靶向治疗,并通过多学科诊疗联盟优化全程管理。唯有打破诊断延迟的桎梏,方能改写Fabry病患者的疾病轨迹。基因治疗等创新技术有望实现“一次治疗、终生治愈”,但当前仍需提升医保覆盖(已纳入中国首批罕见病目录)及多学科诊疗能力。

