Cell Metabolism|初波/孙金鹏/于晓/柴人杰/徐云飞团队合作在靶向铁死亡治疗肝脏疾病物领域获得重大突破
时间:2025-08-25 12:14:34 热度:37.1℃ 作者:网络
2024年10月9日,山东大学基础医学院初波教授研究团队,联合孙金鹏教授团队,于晓教授团队,以及东南大学的柴人杰教授和山东大学齐鲁医院徐云飞教授团队,共同在Cell Metabolism杂志上在线发表了他们最新的研究成果 “Sensing steroid hormone 17a-hydroxypregnenolone by GPR56 enables protection from ferroptosis-induced liver injury”。研究团队鉴定了类固醇激素17a-hydroxypregnenolone 的膜受体GPR56与铁死亡密切相关。

原文链接:https://doi.org/10.1016/j.cmet.2024.09.007
铁死亡(Ferroptosis)是由于过度累积的过氧化磷脂以铁依赖的方式诱发的,是新发现的、不同于细胞凋亡和坏死的一种程序性细胞死亡。谷胱甘肽过氧化物酶 4 (GPX4)、铁死亡抑制蛋白 1 (FSP1)、GTP 环化水解酶1 (GCH1) 或二氢乳清酸脱氢酶 (DHODH) 是重要的可调节非酶或脂氧合酶刺激的过氧化脂质的消除。新的证据表明,铁死亡与缺血再灌注损伤(IRI)、药物诱导的组织损伤有关,抑制铁死亡可减轻相应的症状。
G蛋白偶联受体(GPCRs)是人类基因组中最大的膜蛋白家族,介导80%以上细胞跨膜信号转导,大约是34% 临床药物的直接靶标。在脊椎动物中,GPCR超家族根据序列和结构相似性被划分为五个家族。粘附类GPCRs(aGPCRs)作为GPCRs的第二大家族,在调节细胞功能和介导许多生理过程中发挥着关键作用,因此深入了解 GPCR 调节铁死亡的机制将有助于开发新的临床疗法来治疗相关疾病。
实验结果
01 GPR56/ADGRG1 参与 DOX 诱导的肝脏损伤
为了研究 aGPCR 是否参与铁死亡反应介导的组织损伤,研究人员首先检测了 DOX 处理后小鼠肝脏 aGPCR 的 mRNA 水平,筛选到包括 GPR56、GPR64 和 GPR97 在内的三个 aGPCR 成员在 DOX 处理后迅速升高,而铁死亡的特异性抑制剂Fer-1逆转了这一表型。为了证实这一表型,研究人员又使用了Con A 诱导的铁死亡相关肝损伤模型。结果显示,只有 GPR56 显著增加,而 GPR64 或 GPR97 几乎没有变化。此外,使用铁死亡的特异性抑制剂 Fer-1 或Lipro-1在很大程度上也降低了 GPR56 的蛋白水平。
为了进一步研究GPR56在体内的作用,研究人员给Gpr56fl/fl小鼠注射了肝脏特异性cre-腺病毒(pAAV8-Tbg promoter-EGFP-Cre和Tbg-pm-Cre),在DOX 处理后对小鼠肝脏进行生化指标的检测,免疫组织化学(IHC)4-HNE 染色和铁死亡发生的标志物Ptgs2 的上调表明,敲除 GPR56 会加剧 DOX 诱导的脂质过氧化和铁死亡的发生。此外,肝脏损伤的标志物血清丙氨酸氨基转移酶(ALT)和天门冬氨酸氨基转移酶(AST)显示,GPR56 基因敲除会导致小鼠肝脏更严重的损伤。此外,研究人员还利用Alb-Cre+/-Gpr56fl/fl 小鼠结果表明GPR56的缺失明显加重了DOX诱导的肝脏损伤的症状,而补充Fer-1则可缓解肝脏损伤。总之,上述数据表明,GPR56对抵抗铁死亡反应驱动的肝脏损伤至关重要,GPR56的确改变了铁死亡的可塑性。
02 膜受体 GPR56 是一种新型铁死亡抑制因子
研究人员通过分析CTRP癌症治疗反应数据库的数据,发现 GPR56 与铁死亡诱导剂的耐受性密切相关。为了阐明 GPR56 在铁死亡中的作用,研究人员在对铁死亡敏感的人纤维肉瘤细胞系HT1080中进行了 aGPCR cDNA 文库筛选。结果发现,过表达 GPR56能特异性的保护HT1080细胞免受Erastin和RSL3诱导的铁死亡。
此外,研究人员还敲低及敲除了 HT1080 和肝癌细胞系 HCC-LM3 中的 GPR56进行功能检测,结果显示GPR56 的敲除使这些细胞对铁死亡更加敏感。此外,研究人员分离了小鼠原代肝细胞,发现小鼠肝脏缺乏 GPR56 会显著增强对铁死亡的敏感性。铁死亡的发生伴随着脂质过氧化的升高,研究人员通过 BODIPY-C11 581/591 染色法检测了 GPR56 KO 细胞的脂质过氧化水平。结果如预期显示,GPR56 的缺失明显增加了 RSL3 诱导的脂质过氧化,而在 GPR56 KO 细胞中回转GPR56 则逆转了这一现象。综上这些结果共同阐明了 GPR56 作为铁死亡抑制因子的功能。
03 GPR56 通过下调 CD36 蛋白表达调控脂质代谢并改变对铁死亡的易感性
为了探究GPR56介导的铁死亡抗性的分子机制,研究人员首先探究GPR56是影响铁死亡的典型途径还是非典型途径。结果显示GPR56 的缺失并没有改变这些与铁死亡相关蛋白表达。由于脂质代谢是驱动铁死亡的主要因素,研究人员进行了非靶向脂质组学分析,发现缺乏 GPR56 会显著上调多种脂质,包括携带花生四烯丙基(AA,20:4)和肾上腺酰基(AdA,22:4)脂肪酰基的 PLs,它们会驱动脂质过氧化的易感性。此外,缺乏 GPR56 会显著增加游离 PUFA 的含量。
根据文献报道CD36 对细胞外 PUFA 的吸收至关重要,而 SREBP1、AMPK 和 ACC1参与细胞脂质的生物合成,为探究GPR56会影响哪些脂质代谢相关的基因,研究人员通过WB检测了这些相关基因的蛋白表达。令人惊奇的是,研究人员发现在 GPR56 KO 细胞中,CD36 的蛋白表达明显增加,而参与脂质代谢的基因水平变化不大。此外,在Gpr56fl/fl 和 Alb-Cre+/-Gpr56fl/fl 小鼠的原代肝细胞中观察到了一致的表型。同样,过表达 GPR56 在很大程度上减少了内源性或外源性 CD36。过表达 G12 会加速 GPR56 介导的 CD36 降解,而敲低 G12 则会显著缓解这一现象。有趣的是,研究人员发现蛋白酶体抑制剂(MG132)无法阻断 GPR56 诱导的 CD36 降解。阿洛司他丁(E64D)是一种特异性的强效蛋白酶抑制剂,它参与溶酶体中的蛋白质降解。研究人员发现,E64D 可抑制 CD36 的下调。相比之下,补充 Bafa1 或氯喹(CQ)对抑制自噬体-溶酶体融合没有影响。根据文献报道,自噬和内吞是促进溶酶体中蛋白质降解的两个主要途径。由于 Bafa1 和 CQ 无法挽救 CD36 的回补,研究人员猜测自噬对于 GPR56 介导的 CD36 下调是不可或缺的。事实上,敲除作为自噬关键因素的 ATG7 也无法逆转这种表型。新的证据表明,GPCR 信号转导参与了内吞。与这些发现相一致,研究人员发现 GPR56 促进了 CD36 在 RAB5 或 FYVE-YFP 指示的内质体中定位。作为阳性对照的脂多糖(LPS)也诱导了 CD36 的内体定位。此外,利用 FlAsH-生物发光共振能量转移(FlAsH-BRET)生物传感器进一步量化了 CD36 的细胞转运模式,以监测其招募到早期内体的情况。
研究人员进一步研究了 CD36 的下调是否有助于 GPR56 介导的铁死亡抵抗。在 GPR56 KO 细胞中加入 CD36 的酶抑制剂 sulfo-N-琥珀酰亚胺油酸钠(图中将其命名为 CD36i),可显著减少 RSL3 诱导的细胞死亡。细胞死亡分析表明,在 GPR56 KO 细胞中敲除 CD36 在很大程度上消除了铁死亡。综上所述,这些数据揭示了 GPR56 通过促进内吞-溶酶体途径介导的 CD36 降解,重塑脂质代谢进而影响铁死亡反应的可塑性。
04 GPR56 的激动剂是一种类固醇激素
根据之前报道所知,一些天然或合成化合物已被鉴定为 GPR56 及其下游激活剂,类固醇激素及其衍生物被确认为 aGPCR 的激动剂,例如倍氯米松(BCM)和皮质醇可激活 GPR9740;黄体酮可激活 GPR12641;脱氢表雄酮(DHEA)可激活 GPR64。特别注意的是,类固醇小化学物 3-α-DOG 可激活 GPR56。HEK293 是一种常用的模型细胞系,对 GPCRs 的转染效率较高,且受内源性 GPCRs 的干扰最小,因此非常适合进行 G 蛋白活性检测。研究人员生成了 GPR56-G12、GPR56-G13 和 GPR56-Gq 融合蛋白来研究体外转导特异性分子效率,结果发现与野生型 (WT) GPR56 相比,GPR56-G12 融合蛋白表现出更强的P19诱导的 G12 信号传导能力。
有趣的是,研究人员筛选了总共 43 种类固醇衍生物,发现17-OH PREG 是唯一一种能在 GPR56-β(ΔS)-G12 或 GPR56-β(ΔS)-G13 融合蛋白表达的 HEK293 细胞中激活 G12 和 G13 信号转导的衍生物,其 EC50 值分别为 17.6 ± 1.4 和 35.8 ± 6.5 μM。研究人员进一步发现,TG2、P19 和 17-OH PREG 可促进 G12 的解离。这些数据表明 TG2 通过 17-OH PREG 对 GPR56 的激活起着积极的异构调节作用,而 P19 与 17-OH PREG 竞争结合到 GPR56 上。事实上,deguelin 以竞争方式抑制了 17-OH PREG 诱导的 GPR56 激活,EC50 值为 350 ± 60 nM。总之,研究人员发现类固醇激素 17-OH PREG 是 GPR56 的激动剂。
配体在aGPCR 的 7TM 束内结合通常会引起细胞外构象变化,这种变化可通过 FlAsH-BRET 分析法检测到。因此,研究人员用这种方法研究了 17-OH PREG 诱导的 GPR56 细胞外环(ECLs)或细胞内环(ICLs)的构象变化。研究人员筛选了分别在 GPR56-ECL-FlAsH 或 GPR56-ICL-FlAsH 构建物的 N 端或 C 端携带 FlAsH 基序(四半胱氨酸 [TC]-tag, CCPGCC)和 Nluc 分子的 GPR56 受体的每个 ECL 或 ICL 的特定位置。在 17-OH PREG 的刺激下,Nluc 标记的 N 端与 FlAsH 标记的 ECL1 和 ECL2 之间的 BRET 信号显著减少,但对 ECL3 的影响很小,这表明 17-OH PREG 可促进 ECL1 和 ECL2 从 GPR56 的 N 端分离。综上所述,这些数据表明 17-OH PREG 能特异性调节 GPR56 细胞内和细胞外区域的构象变化。
05 17-OH PREG 可以诱导GPR56 构象发生变化
研究人员利用拉马克遗传算法(LGA)和 Auto Dock 4 进行了分子动力学模拟(MD),探讨了 17-OH PREG 与 GPR56 配体口袋的结合模式。研究人员以皮质醇-GPR97 结构为模板,通过瑞士模型生成了 GPR56 结构模型,因为 GPR97 的结构与 GPR56 具有高度的序列同源性。来自 TM3、TM5-TM7 和 ECL2 的 7 个疏水残基和 3 个极性残基组成了潜在的 17-OH PREG 结合口袋。将 17-OH PREG 口袋残基突变为丙氨酸会削弱激动剂诱导的 G12 信号传导,而随机选择的另外两个周围残基 L4903.44A 和 V556ECL2A 则没有显著影响。接下来,研究人员使用基于 FlAsH-BRET 的 GPR56 传感器来监测 17-OH PREG 结合袋中突变的影响。L4823.36A、L4865.51A、W563ECL2A、S5705.35A、S5705.35V、Y5715.36F、N5745.39L、W6236.53A 和 F6437.42A 的突变显著影响了 17-OH PREG 诱导的 ECL2 构象变化。GPR56 基因突变与脑部疾病相关,COSMIC 人类疾病数据库中存在多个 GPR56 错义突变,包括 L4492.53del、S4853.39P、L4873.41P、E4963.50K、R565ECL2W 和 L6407.39W。有趣的是,除 S485P 外,几乎所有突变都失去了 G12 激活能力和抗铁死亡能力。
06 17-OH PREG 通过激活 GPR56 保护细胞免于铁死亡
上述数据促使研究人员想进一步研究 17-OH PREG 是否通过 GPR56 发挥抗铁死亡作用。对类固醇代谢物的筛选后发现,17-OH PREG 能明显抑制RSL3诱导的铁死亡,而除了 17β-estradiol 外,其他化合物都没有明显的效果。此外,流式结果显示17-OH PREG 还能有效消除脂质过氧化的发生。
此外,17-OH PREG 无法挽救HT1080 GPR56 KO 细胞的铁死亡。相反,17β-雌二醇在很大程度上抑制了铁死亡,表明 17β-雌二醇介导的铁死亡抵抗与 GPR56 无关。此外,过表达 GPR56 可恢复 17-OH PREG 对 GPR56 缺失的 HT1080 细胞的影响。总之,这些数据揭示了 GPR56 在 17-OH PREG 介导的铁死亡反应中不可或缺。
07 17-OH PREG 通过 GPR56 减轻 DOX、IR 或 MCDD 诱导的肝损伤
为探索 17-OH PREG 在体内的生理特性,研究人员继续使用了 DOX诱导的肝损伤模型。如预期所想,铁死亡特异性抑制剂Fer-1 能有效消除WT 或 GPR56 KO 小鼠肝脏的 DOX 引起的损伤症状。此外,IHC 染色显示,给小鼠注射 17-OH PREG 可降低脂质过氧化反应,并减轻WT小鼠肝脏的损伤。然而,在小鼠肝脏特异性缺失 GPR56 的情况下,17-OH PREG 的保护作用基本消失。研究人员还采用了缺血再灌注诱导的肝损伤模型,并在 Gpr56fl/fl 和 Alb-Cre+/- Gpr56fl/fl 小鼠中取得了相同的结果,这表明 17-OH PREG 对铁死亡相关的组织损伤具有保护作用。因此,研究人员研究了 17-OH PREG 对蛋氨酸胆碱缺乏饮食(MCDD)诱导的 MASH 模型的影响。不出所料,H&E 和油红 O 染色显示 MCDD 促进了气球变性和肝细胞脂肪变性,这是 MASH 的标志。此外,研究人员还在 MCDD 模型中观察到严重的肝损伤,给予 17-OH PREG 治疗可显著延缓 MASH肝损伤的进展。
此外,研究人员还采集了 MAFLD 患者的血清和肝脏样本,并检测了 GPR56/17-OH PREG-CD36 轴的变化。结果表明,在 MAFLD 患者中,17-OH PREG 的丰度显著升高。这些数据共同表明,GPR56/17-OH PREG-CD36 轴参与了 MASH 的进展,从临床转化的角度看具有重要意义。
本成果突出贡献
本成果突出贡献
(1)GPR56 是一种有效的铁死亡抑制因子
(2)GPR56 通过下调 CD36 重塑脂质代谢以抵抗铁死亡反应
(3)确定 17-OH PREG 是 GPR56 的强效激动剂,可抑制铁死亡反应
(4)靶向 GPR56-17-OH PREG-CD36 轴有助于缓解铁死亡反应诱导的肝损伤
综上所述,该研究团队阐明了类固醇代谢物17-OH PREG通过GPR56/ADGRG1途径抵抗铁死亡的新分子机制,并首次揭示了GPCR和相应的信号传导调控的铁死亡在临床治疗中的潜在重要作用。研究结果为肝脏损伤的潜在治疗提供了新的见解,深度剖析了GPCR受体与铁死亡之间的紧密联系,从临床角度来看,通过治疗性服用 17-OH PREG 治疗肝病可有效预防急性和慢性肝功能衰竭,为临床铁死亡相关疾病的治疗方法的开发和应用提供了新的研究策略。
专家点评
王福俤 教授(浙江大学求是特聘教授,新乡医学院校长/南华大学副校长)
初波/孙金鹏/于晓/柴人杰/徐云飞等《Cell Metabolism》合作成果提供了坚实实验数据,揭示了G 蛋白偶联受体GPR56可感知类固醇激素17α-羟基孕烯醇酮(17-OH PREG)进而保护阿霉素(DOX)或缺血再灌注(IR)诱发肝损伤新效应和新机制。G 蛋白偶联受体(GPCR)是数量最多的细胞表面受体。铁死亡是铁依赖的脂质过氧化驱动的程序性细胞死亡方式,它与缺血再灌注损伤、阿霉素诱发组织损伤等多种病症相关。深入探究 GPCR 调节铁死亡机制将有助于发现疾病防治新策略。初波教授等这项成果阐明了铁死亡与GPCR受体之间调控新机制,为肝脏损伤相关疾病治疗提供了潜在新靶点。
研究团队首先在小鼠肝损伤模型中,发现GPR56显著上调。进一步通过 GPCR cDNA 文库筛选,发现 GPR56改变了铁死亡的可塑性。这项研究首次证明了由 GPR56/ADGRG1 激活介导的跨膜信号传导可有效抑制铁死亡,这表明 GPCRs 和相应的信号传导在调节铁死亡反应和组织损伤中发挥着重要作用。
作者首次发现17-OH PREG 是GPR56/ADGRG1 的配体,它能增强 GPR56 的抗铁死亡效应,进而减轻肝脏受损前后的急性和慢性损伤。对于铁死亡相关的肝脏损伤疾病来说,类固醇激素17-OH PREG给药可能是一种新型、安全有效治疗方式,这项研究重塑了人们对类固醇激素作用的认知。
该成果还揭示了GPR56-CD36 轴介导的脂质代谢是抗铁死亡新途径,GPR56通过促进CD36的内吞-溶酶体降解途径抑制铁死亡,其介导的信号转导是维持肝脏稳态的必要条件,为肝脏损伤中的临床试验提供了理论依据。
专家点评
李小英 教授(复旦大学)
源自肾上腺的类固醇激素在许多生理系统中发挥着不可或缺的作用,包括生长、生殖、炎症和代谢。然而一些具有生物活性的类固醇激素前体物质(比如17羟基孕酮,17羟基孕烯醇酮等)在代谢性疾病中的作用尚不清楚。我们在2020年首次发现血浆17羟基孕酮水平升高与2型糖尿病相关,同时鉴定出17羟基孕酮的关键合成酶17羟化酶(CYP17A1)是2型糖尿病中的新型干预靶点,靶向CYP17A1开发出肝脏选择性和特异性的抑制剂有望为2型糖尿病的防控带来新的希望(JCI 2020)。2022年孙金鹏教授和于晓教授团队鉴定了孕酮和17羟基孕酮的膜受体GPR126(Nat Chem Biol 2022, PNAS 2022),证明了这两种激素通过GPR126调控三阴性乳腺癌的发展,完善了甾体激素作用的非基因组机制学说。
临床上肝损伤十分常见,包括病毒性、药物性、自身免疫性以及代谢相关脂肪性肝炎(MASH),铁死亡参与多种病因的肝损伤,尤其MASH。在本文中,初波教授与孙金鹏教授、于晓教授等团队通力合作,首次证明由粘附类G蛋白偶联受体GPR56/ADGRG1激活介导的跨膜信号可以有效地抑制铁死亡,表明GPCRs及其信号在调节铁死亡和组织损伤中的重要作用。他们鉴定出一种类固醇激素前体17羟基孕烯醇酮(17-OH PREG)是GPR56的新配体,可增强GPR56的抗铁死亡作用,补充17-OH PREG可有效减轻急性和慢性肝损伤。
他们的研究首次证明铁死亡可以通过跨膜受体依赖的方式调控,即GPCRs作为细胞外感知和细胞间通讯的核心调控者发挥铁死亡的调节作用。他们的研究从临床上证明了17-OH PREG与代谢相关脂肪性肝病(MAFLD)的相关性,其疾病进展也与17-OH PREG/GPR56和CD36轴可能相关。因此,他们提出通过治疗性使用17-OH PREG极可能有效地预防急性和慢性肝衰竭,治疗最为常见的肝损伤性疾病MASH。他们的这些研究发现,打开了探索粘附类GPCR蛋白在代谢性疾病调控作用的新篇章。
专家点评
刘军力 教授(上海交通大学)
类固醇激素作为生物化学领域中一类关键的生物活性分子,其在维持生物体内环境稳定、调节糖脂代谢以及促进生长发育等方面扮演着十分重要的角色。普遍认为,这些激素会进入靶细胞并与细胞质或细胞核中的特定受体结合,进而调控基因的转录过程,而这一过程通常需要数小时甚至数天的时间。然而,随着研究的深入,越来越多的证据表明类固醇激素还具有非基因组效应,它们能够通过与细胞膜上的受体结合,发挥快速的生物学作用。尽管已有研究揭示了极少数类固醇激素(如雌激素)的细胞膜受体,但大多数类固醇激素的膜受体尚未被确切鉴定,其快速作用的机制仍然是一个未解之谜。在这一领域,孙金鹏教授团队和于晓教授团队在过去五年中取得了一系列突破性进展,他们鉴定了多个类固醇激素的G蛋白偶联受体(GPCR)膜受体。具体来说,他们发现了粘附类受体GPR97(ADGRG3)是糖皮质激素的膜受体;鉴定了粘附类受体ADGRG2是类雄激素脱氢表雄酮的膜受体;此外,他们还揭示了孕酮和17羟基孕酮是粘附类受体GPR126(ADGRG6)的内源性配体。这些发现表明,粘附类GPCR构成了识别类固醇激素的一个GPCR亚家族。
近日,初波教授、孙金鹏教授和于晓教授等研究团队又取得了新的研究成果,他们发现内源性类固醇激素17羟基孕烯醇酮能够激活粘附类受体GPR56,从而抵抗细胞铁死亡,并有效减轻损伤前或损伤后的肝损伤。这一发现揭示了17-OH PREG-GPR56轴介导的信号转导作为一种新的抗铁死亡途径,对于维持肝脏稳态具有重要意义,并为肝损伤的潜在治疗提供了新的思路。这一系列在类固醇激素GPCR受体领域的开创性工作,不仅为类固醇激素非基因组机制学说提供了重要的补充,而且进一步拓展和完善了我们对类固醇激素-膜受体信号通路在不同生理和病理生理过程中作用的理解。这些研究成果不仅在基础生物学领域具有深远的意义,也为未来的临床应用提供了新的方向。
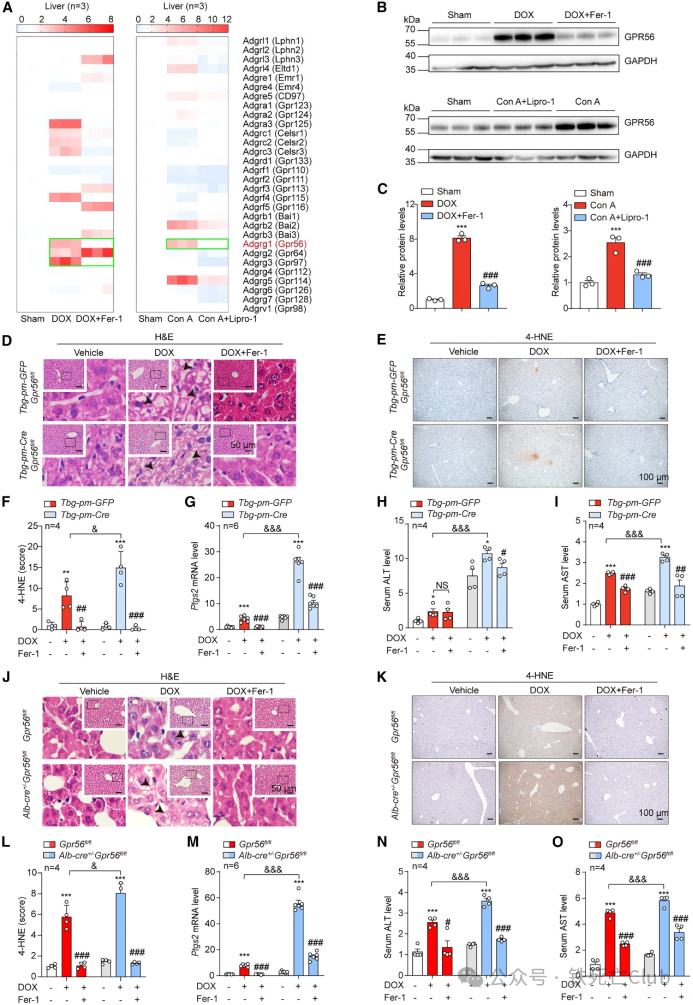
图1. GPR56/ADGRG1 参与 DOX 诱导的肝脏损伤
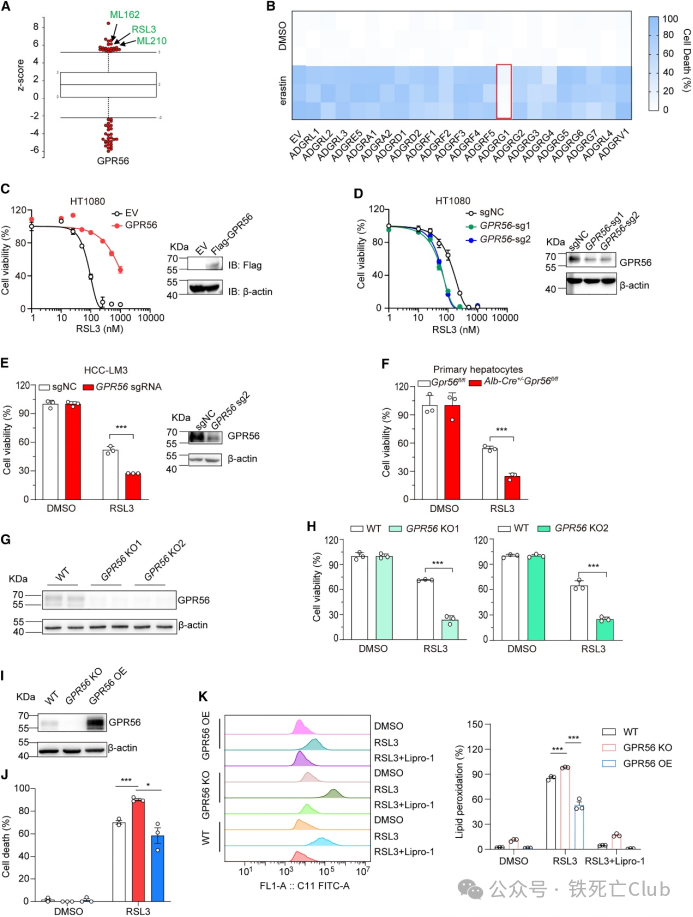
图2. 膜受体 GPR56 是一种新型铁死亡抑制因子
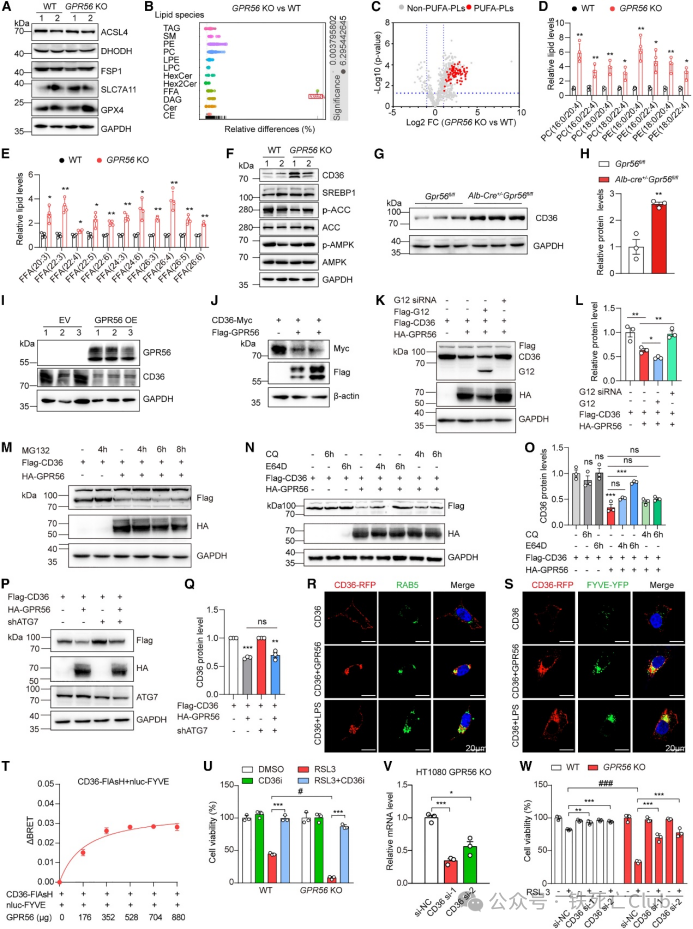
图 3. GPR56 通过下调 CD36 蛋白表达调控脂质代谢并改变对铁死亡的易感性
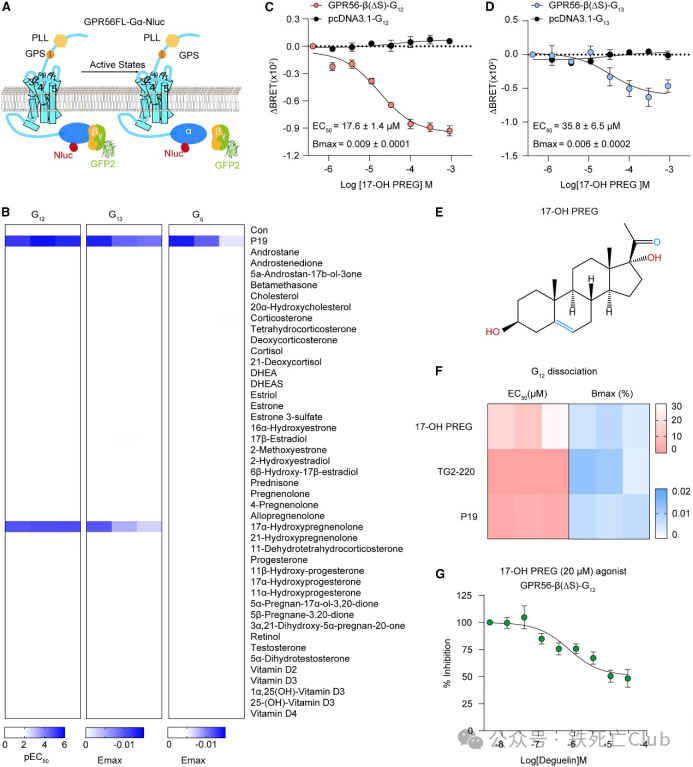
图4. GPR56 的激动剂是一种类固醇激素
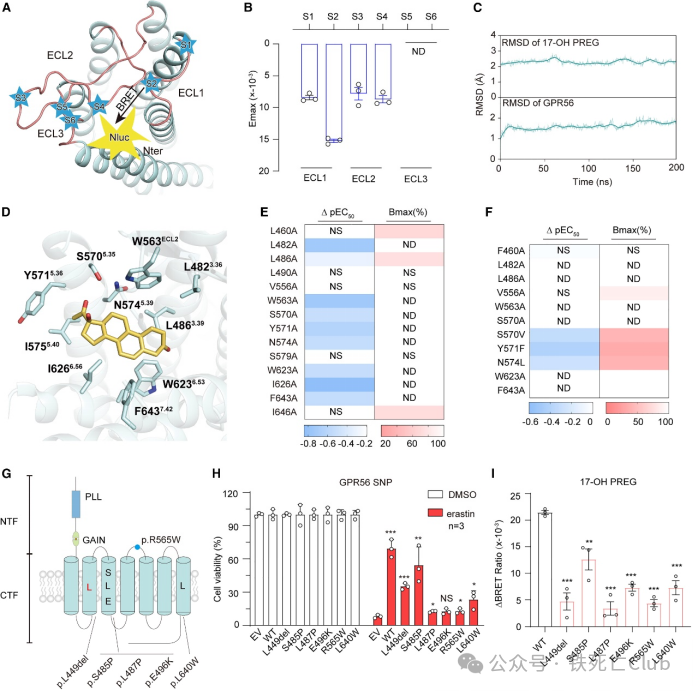
图5. 17-OH PREG 可以诱导GPR56 构象发生变化
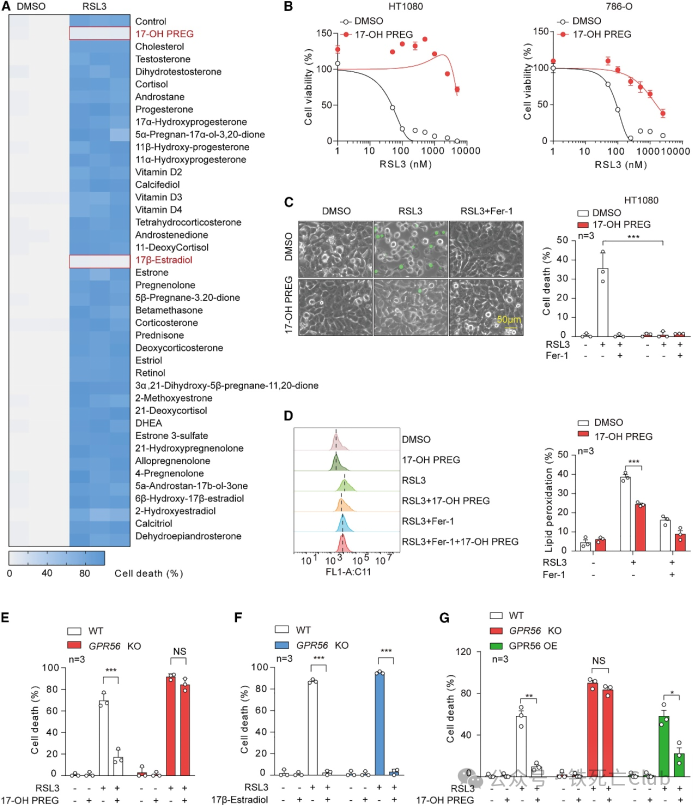
图6. 17-OH PREG 通过激活 GPR56 保护细胞免于铁死亡
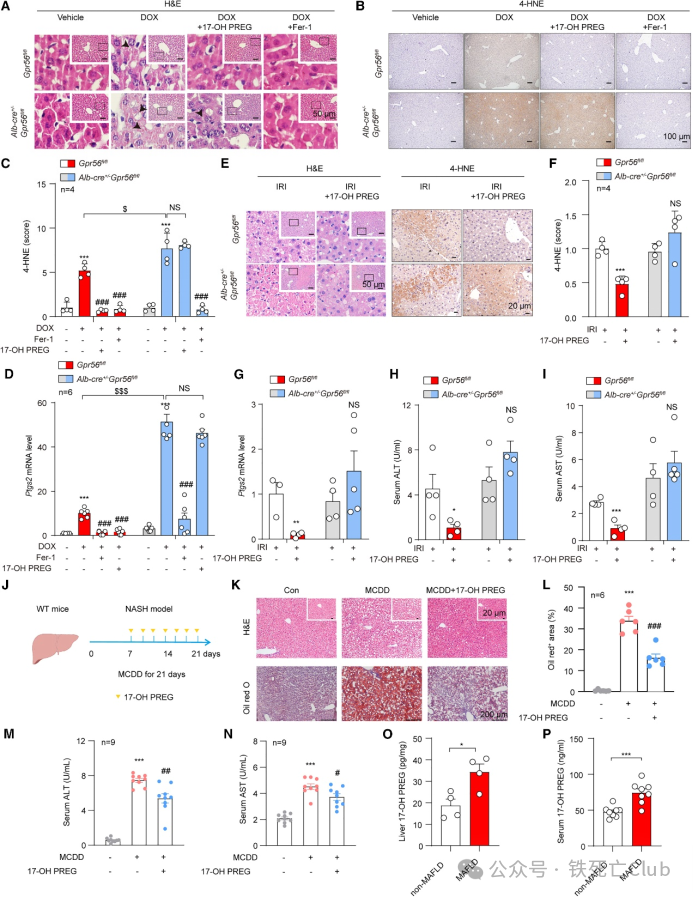
图7. 17-OH PREG 通过 GPR56 减轻 DOX、IR 或 MCDD 诱导的肝损伤

