特别关注|诱导铁死亡: 逆转胰腺癌对吉西他滨化疗耐药性的新策略
时间:2025-09-07 12:08:36 热度:37.1℃ 作者:网络
胰腺癌是一种消化系统恶性肿瘤,具有早期诊断难、治疗成功率低、预后差等特点。胰腺导管癌(PDAC)是最常见的胰腺癌类型,占所有胰腺恶性肿瘤的95%以上,5年生存率仅为13%,其死亡率在所有实体瘤中最高。化疗是目前提高胰腺癌患者生存率的主要治疗手段,吉西他滨(GEM)是化疗的基本药物之一,但其疗效常因耐药性而受限。已有研究证实,诱导肿瘤细胞铁死亡不仅能抑制肿瘤细胞生长,还能逆转化疗耐药性。不同于细胞凋亡、细胞坏死或细胞自噬,铁死亡主要是由胞内铁离子与活性氧(ROS)积累所致,其发生的标志为细胞膜特定脂质发生过氧化。研究表明,通过诱导铁死亡来改善胰腺癌细胞的GEM耐药性,是一种潜在的新型治疗策略。因此,深入研究铁死亡与GEM耐药机制之间的关系,对改善胰腺癌患者的治疗效果具有重要意义。
1胰腺癌的GEM耐药机制
PDAC的术后复发率超过50%,5年生存率约为28%,中位生存时间仅18个月。术后辅助化疗能显著提高生存率并降低复发率。GEM通过影响DNA合成来杀伤肿瘤细胞,是PDAC患者化疗的常用药物之一。然而,胰腺癌细胞会对GEM逐渐产生耐药性,导致其疗效减弱。GEM的耐药机制可以分为细胞内源性机制和非细胞自主耐药机制。
通过肿瘤细胞内部修饰产生的化疗耐药机制被称为细胞内源性耐药机制,涉及代谢、表观遗传重编程和上皮-间质转化(EMT)等方面。例如,GEM的转运需借助胰腺癌细胞上的NTs(核苷转运蛋白),而hENT1(人平衡型核苷转运蛋白1)表达降低会阻碍该进程;脱氧胞苷激酶(dCK)缺乏会影响GEM活化,促进耐药;RRM1(核糖核苷酸还原酶亚基M1)高表达会增强核糖核苷酸还原酶活性,减少GEM的DNA掺入;CDA(胞苷脱氨酶)将GEM转化为吉西他滨二磷酸,阻止其激活;EMT在增强肿瘤侵袭性与化疗耐药中均起到关键作用。
非细胞自主耐药机制主要由肿瘤微环境的成分驱动。肿瘤相关成纤维细胞(CAF)形成的细胞外基质物理屏障,会阻碍药物的灌注输送,并引起组织缺氧,影响GEM的有效性;此外CAF分泌竞争性的脱氧胞苷也参与耐药。肿瘤相关巨噬细胞通过上调CDA表达和竞争性抑制GEM激活,进一步推动耐药。其他如细菌、神经元和癌症相关脂肪细胞等也与GEM耐药相关。
综上所述,胰腺癌细胞通过细胞内源性与非细胞自主耐药机制对GEM产生耐药。铁死亡过程中,细胞膜脂质过氧化产物的累积会触发肿瘤细胞死亡。胰腺癌细胞通过多种机制,如提高谷胱甘肽(GSH)和谷胱甘肽过氧化物酶4(GPX4)活性,抵抗铁死亡引起的氧化应激作用,与GEM的耐药密切相关。化疗药物和铁死亡诱导剂联合使用的方案可能是逆转胰腺癌对GEM耐药并增强化疗效果的潜在策略。靶向铁死亡过程的关键组分和调节因子,如增强多不饱和脂肪酸(PUFA)的过氧化、破坏肿瘤细胞的抗氧化防御系统,以及调节铁代谢,都可以提升胰腺癌细胞对GEM的敏感性,进而克服耐药机制对胰腺癌中GEM疗效的影响。
2相关的细胞内源性GEM耐药机制
胰腺癌细胞中,铁死亡细胞内源性GEM耐药机制涵盖了转录调控、代谢物质转运和蛋白酶降解等多方面,本文将其归纳为PUFA过氧化、铁代谢调控和抗氧化系统3条途径。PUFA过氧化涉及铁死亡关键底物的产生和重塑过程;铁代谢调控揭示了铁离子水平对铁死亡的影响;而抗氧化系统阐述了针对脂质过氧化的内源性防御如何发挥作用。
2.1 通过增强PUFA过氧化抑制胰腺癌细胞的GEM耐药性
脂质过氧化反应是细胞铁死亡发生的重要因素。由于分子结构中含有更多的双键,PUFA比单不饱和脂肪酸(MUFA)或饱和脂肪酸更容易被氧化,是脂质过氧化的主要底物。研究发现,亚油酸和α-亚麻酸可以部分诱导GEM耐药的胰腺癌细胞发生铁死亡,进而抑制肿瘤生长,证明了PUFA积累可以通过促进细胞铁死亡降低胰腺癌中的GEM耐药性。在PUFA过氧化过程中,首先长链脂酰辅酶A连接酶4(ACSL4)将PUFA激活为PUFA-CoA(不饱和脂肪酸辅酶A酯),随后LPCAT3(溶血磷脂酰胆碱酰基转移酶3)通过磷脂重塑将PUFA-CoA整合到细胞膜的磷脂结构中,形成PUFA-PL(多不饱和脂肪酸磷脂),进而在亚铁离子和自由基的共同作用下诱导细胞铁死亡。ADP核糖基化因子6(ARF6)与Hippo-YAP及Hippo-YY1通路均可以提高ACSL4表达量,增强PUFA过氧化进程。同时,抑制硬脂酰辅酶A去饱和酶1(SCD1)对PUFA与MUFA比例的调节作用也可以增强PUFA过氧化(图1)。
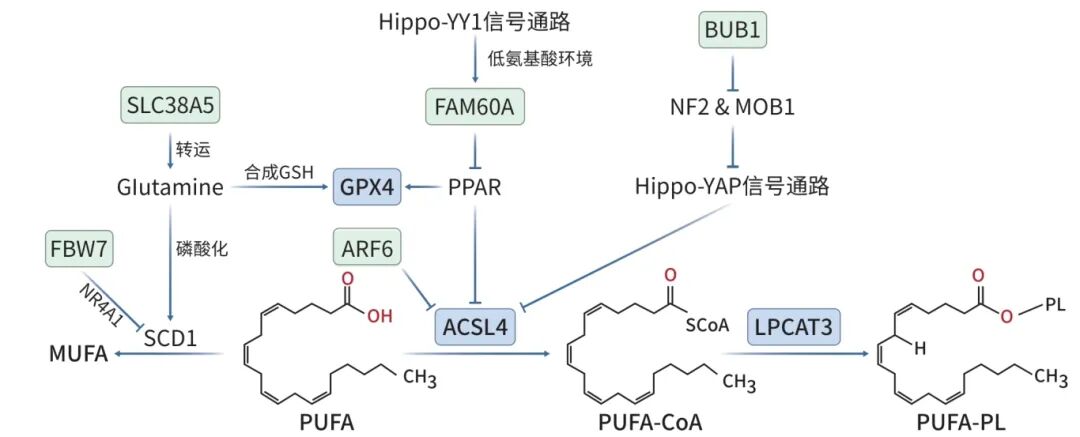
注: SLC38A5,溶质载体家族35成员5;Glutamine,谷氨酰胺;FBW7,F框/WD重复结构域蛋白7;NR4A1,核受体亚家族4A组;FAM60A,序列相似度60家族成员A;PPAR,过氧化物酶体增殖物激活受体;ACSL4,长链脂酰辅酶A连接酶4;ARF6,ADP核糖基化因子6;NF2,神经纤维蛋白2;BUB1,苯并咪唑出芽抑制解除同源物蛋白1;MOB1,Mps-结合者激酶激活因子样1。
图1 PUFA过氧化调节通路
ARF6是RAS家族中一种促进胰腺癌细胞增殖的重要因子,与铁死亡和GEM耐药具有相关性。研究发现,敲除ARF6可以上调ACSL4转录翻译后的水平,使胰腺癌细胞处于更容易受氧化应激影响的状态,进而提高铁死亡诱导剂对PDAC细胞铁死亡的诱导效果,并抑制PDAC细胞的GEM耐药性。同时,GEM相关的代谢蛋白dCK和hENT1表达水平上调可能也与耐药性受抑制有关。因此,通过ARF6调控ACSL4介导的脂质过氧化可能是改善胰腺癌GEM耐药性的新思路。
Hippo-YAP信号通路中的YAP(Yes相关蛋白)可以通过调节相关基因的表达增强细胞对铁死亡的敏感性,例如ACSL4与转铁蛋白受体1(TFR1)。BUB1(苯并咪唑出芽抑制解除同源物蛋白1)是一种与胰腺癌患者总体生存率呈负相关的丝裂检查点激酶。研究发现,通过在GEM耐药的胰腺癌细胞中敲除BUB1或对其使用铁死亡诱导剂处理,可以增强细胞内NF2和MOB激酶激活物1对Hippo-YAP信号通路的抑制作用,升高ACSL4等铁死亡相关基因的表达水平,从而促进胰腺癌细胞的铁死亡并增强GEM的细胞毒性作用。
此外,Hippo-YY1信号通路也可以调节相关基因的表达诱导铁死亡发生。研究发现,Hippo-YY1信号通路在低氨基酸环境中会被激活,并启动FAM60A的转录。FAM60A通过抑制PPAR,可以降低ACSL1/4并提高GPX4的表达水平,进而抑制PDAC细胞的铁死亡。该研究发现,敲除FAM60A以诱导胰腺癌细胞铁死亡可以有效抑制肿瘤生长,并增强肿瘤细胞对GEM的化疗敏感性。
SCD1可以催化饱合脂肪酸转化为MUFA,调节细胞膜中MUFA与PUFA的比例,间接影响脂质过氧化和铁死亡水平。氨基酸转运蛋白SLC38A5转运的谷氨酰胺通过两种途径影响胰腺癌的GEM耐药性。在磷酸化途径中,谷氨酰胺通过磷酸化作用可以触发肿瘤细胞内的mTOR-SREBP1-SCD1通路,将PUFA转化为MUFA,抑制铁死亡。同时,谷氨酰胺是GSH的合成底物之一,GSH与GPX4共同作用可以降低细胞内的ROS水平并抑制铁死亡。因此,靶向并抑制SLC38A5可以诱导GEM耐药的胰腺癌细胞铁死亡,抑制了肿瘤的生长和转移。另有研究通过铁死亡抑制剂铁抑素-1与凋亡抑制剂z-VAD-FMK处理FBW7过表达的细胞,证明E3泛素连接酶复合物的关键组分FBW7可以通过NR4A1转录因子抑制SCD1表达,减少油酸合成和CoQ10生成,提高脂质过氧化水平并诱导细胞发生铁死亡和凋亡,进而增强GEM的细胞毒性。FBW7调控NR4A1的具体机制还有待于进一步研究。许多针对NR4A与SCD1的抑制剂已经在开发中,将会为化疗干预提供新的靶点。
2.2 调控铁代谢改善胰腺癌细胞的GEM耐药性
PUFA与膜结合后,不稳定铁库(LIP)中的亚铁离子通过芬顿反应产生高活性的羟自由基,这些自由基将PUFA-PL进一步氧化成磷脂氢过氧化物(PUFA-PL-OOH)。细胞内PUFA-PL-OOH引发链式反应并不断积累,最终导致铁死亡发生(图2)。TFR1是铁代谢过程中的重要蛋白,通过与转铁蛋白结合,可以经胞吞作用将铁输送到内体中。内体的酸性环境使铁从转铁蛋白中释放,并通过二价金属转运蛋白1转运至细胞质,形成LIP。前文中的研究通过敲除BUB1或用铁死亡诱导剂处理可以抑制GEM耐药的胰腺癌细胞中的Hippo-YAP信号通路,升高TFR1表达量,进一步增加LIP丰度,从而促进细胞铁死亡。
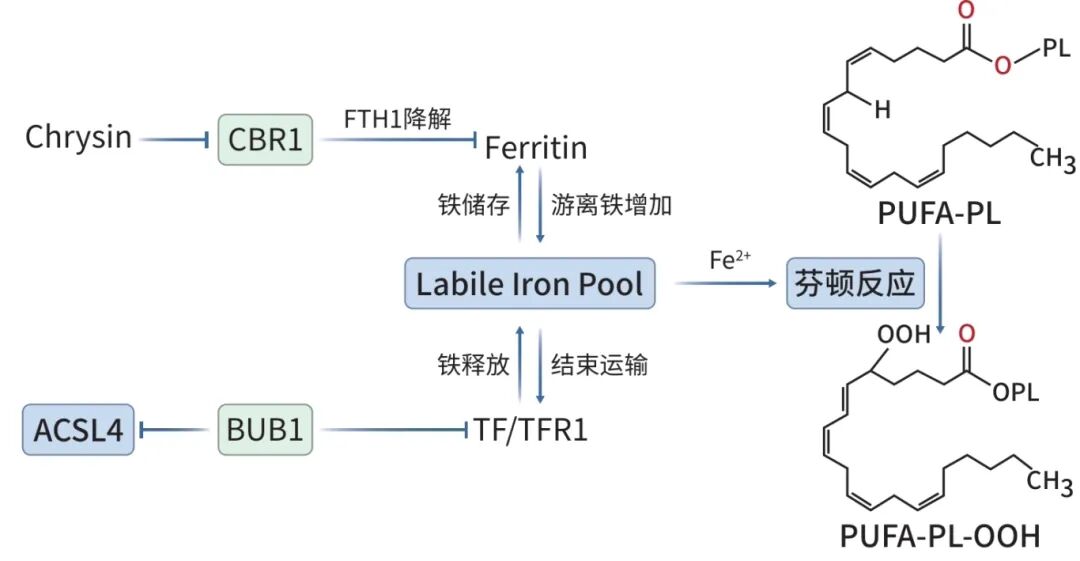
注: FTH1,铁蛋白重链多肽1;TF,转铁蛋白;TFR1,转铁蛋白受体1;Ferritin,铁蛋白;Labile Iron Pool,不稳定铁库;CBR1,碳酮还原酶1;Chrysin,白杨素。
图2 铁代谢调节通路
铁蛋白是一种由重链铁蛋白和轻链铁蛋白两种亚基组成的铁储存蛋白复合体,在细胞内铁平衡中发挥重要作用,且其可以在自噬作用下被分解,改变LIP丰度,进而影响铁死亡的敏感性。CBR1是一种与肿瘤进展和化疗耐药性密切相关的NADPH依赖性酶,在胰腺癌组织中高表达。研究发现作为CBR1抑制剂的黄酮类化合物白杨素直接与胰腺癌细胞内的CBR1结合,可以抑制其活性,增加细胞内的ROS水平,并导致ROS依赖性的自噬作用。该研究进一步发现,这种自噬作用会导致重链铁蛋白降解和细胞内游离铁增加,进而通过芬顿反应促进胰腺癌细胞铁死亡,并增强胰腺癌细胞对GEM的敏感性。
2.3 靶向抗氧化系统减弱胰腺癌细胞的GEM耐药性
细胞膜ROS过度积累会使细胞膜完整性受损,最终导致细胞铁死亡发生,而抗氧化系统在其中发挥重要的调节作用。细胞膜上的谷氨酸/胱氨酸逆向转运体(System XC-)可以将细胞外的胱氨酸转运至细胞内,在TXNRD1的催化下还原为半胱氨酸,参与GSH的合成。GSH是GPX4的一种辅因子,两者共同作用可以将以PUFA-PL-OOH为主的ROS还原为PUFA-PL-OH(多不饱和脂肪酸磷脂醇)等脂质醇形式,抑制ROS积累,防止细胞膜结构的完整性被破坏,从而阻止铁死亡的发生。研究发现通过以下多个因子或进程可以影响GPX4的表达水平或降解机制,进而抑制System XC--GSH-GPX4介导的抗氧化系统,并改善胰腺癌细胞的GEM耐药(图3)。
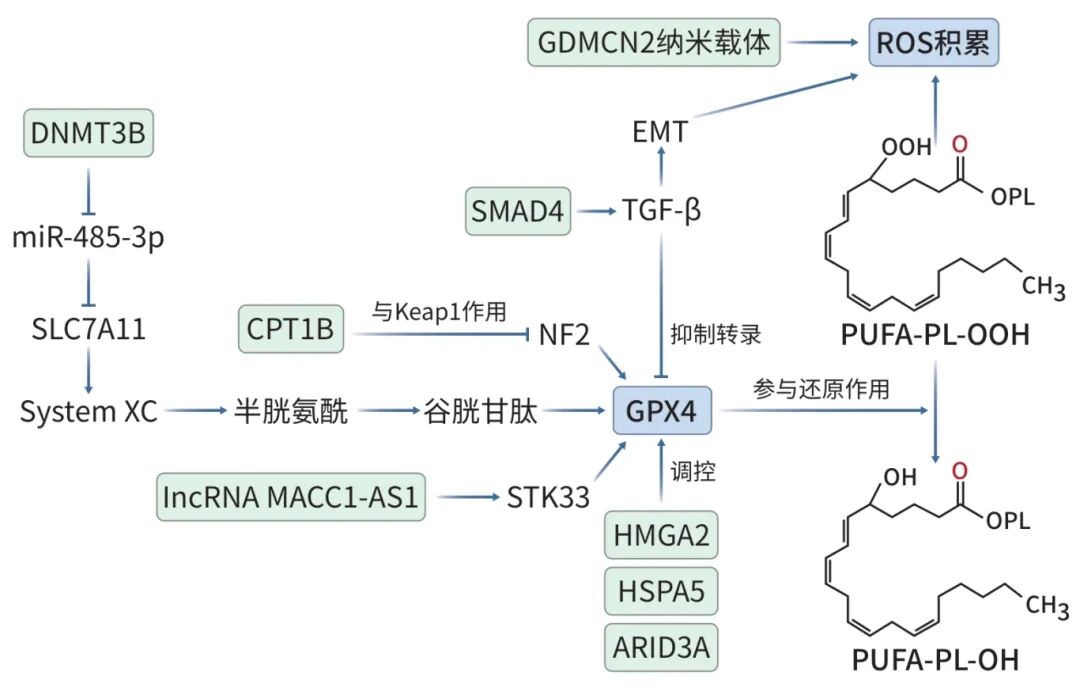
注: DNMT3B,DNA甲基转移酶3B;SLC7A11,溶质载体家族7成员11;CPT1B,肉碱棕榈酰转移酶1(CPT1)家族亚型;SMAD4,SMAD家族成员4;lncRNA MACC1-AS1,MACC1反义RNA1;STK33,丝氨酸/苏氨酸激酶33;HSPA5,热休克蛋白家族A成员5;GDMCN2,吉西他滨-DVDMS@金属有机框架@共价有机框架-NRP2抗体;ARID3A,AT丰富结构域相互作用蛋白3A。
图3 抗氧化系统调节通路
核红细胞2相关因子2(NRF2)是一种致癌的转录因子。ROS水平升高时,Keap1与NRF2结合并促其泛素化降解的能力减弱,导致NRF2水平升高并与细胞核中ARE基因结合,启动System XC-和GPX4等铁死亡相关的基因转录,抑制铁死亡。有研究发现CPT1B可以与Keap1相互作用调节NRF2的水平。敲除CPT1后,GPX4表达水平降低,从而导致ROS积累并诱导铁死亡。该研究进一步通过体外和体内实验证明,针对CPT1B来抑制GPX4介导的抗氧化系统并诱导细胞铁死亡,可以显著增强GEM对化疗耐药的PDAC细胞的疗效。
高迁移率蛋白A2(HMGA2)是一种胰腺癌中高表达的致癌基因。研究发现,胰腺癌细胞中HMGA2可以通过pK4甲基化和pK27乙酰化来提高增强子活性,并激活mTORC1-4EBP1和mTORC1-S6K信号轴,促进GPX4蛋白的合成,从而抑制细胞铁死亡的发生。该研究进一步发现,敲除HMGA2并抑制GPX4介导的抗氧化系统,可以显著提高胰腺癌细胞对GEM与铁死亡诱导剂RAS选择性致死分子3(RSL3)联合使用的效果。
lncRNA MACC1-AS1在多种癌症中均与耐药性有关。最近基于胰腺癌的研究发现,lncRNA MACC1-AS1在GEM耐药的胰腺癌细胞中高表达,且与患者不良预后相关。lncRNA MACC1-AS1与蛋白激酶STK33的相互作用可以抑制STK33的泛素化降解,导致STK33在细胞内积累,进而阻止GPX4蛋白降解,抑制铁死亡,并促进胰腺癌细胞对GEM的耐药。针对其开发诱导铁死亡的临床治疗方法也是一种可能策略。
HSPA5是一种主要表达在内质网中的分子伴侣蛋白,其表达水平与PDAC患者的不良预后密切相关。研究发现,在激活转录因子4的诱导下,HSPA5通过与GPX4结合可以阻止GPX4降解,抑制脂质过氧化,进而抑制细胞铁死亡与GEM的细胞毒活性,影响患者预后。而使用RNAi、儿茶素或柳氮磺胺吡啶抑制HSPA5对GPX4的作用,可以在体外和小鼠胰腺癌动物模型中增强PDAC细胞对GEM的敏感性。
肿瘤细胞的EMT不仅与肿瘤的发生、侵袭、转移和耐药性相关,而且会提高细胞对铁死亡的敏感性。SMAD4是一种针对胰腺癌的抑癌基因,可以通过诱导细胞EMT增强铁死亡敏感性。研究发现,在胰腺癌中,SMAD4可以影响转化生长因子β1对EMT方向的诱导,提高细胞脂质过氧化水平,并通过结合GPX4启动子抑制GPX4的转录与表达,促进肿瘤细胞铁死亡。研究人员运用这一特性,使用铁死亡诱导剂RSL3联合GEM有效杀伤SMAD4阳性的PDAC细胞,显著增强了疗效。
3铁死亡相关的非细胞自主性GEM耐药机制
胰腺癌的基质细胞和肿瘤微环境在铁死亡相关的非细胞自主GEM耐药机制中同样发挥重要作用,并主要通过调节抗氧化系统途径诱导铁死亡,进而改善胰腺癌的GEM耐药(图3)。
先前的研究已发现肿瘤微环境中的CAF与免疫细胞可以通过分泌小分子(microRNA、lncRNA等)或增强细胞外基质的致密性促进肿瘤细胞的化疗耐药性。最近的研究发现在GEM耐药的胰腺癌肿瘤微环境中,基质细胞(CAF与免疫细胞)的ARID3A(AT丰富结合域3A)表达水平显著升高,且与化疗耐药性相关。PTEN可以增强PDAC细胞对GEM的敏感性,而ARID3A可以与PTEN启动子区域结合并降低其转录活性,减弱PTEN对PI3K/Akt/mTOR通路的抑制作用,同时提高GPX4的表达水平,进而抑制铁死亡。该研究发现,肿瘤基质细胞中ARID3A对PDAC细胞铁死亡的抑制作用增强了细胞的化疗耐药性,而抑制ARID3A从而诱导铁死亡为研究改善胰腺癌化疗耐药性提供了新思路。
PDAC的肿瘤微环境中经常由于血管生成障碍、细胞异常增殖与基质纤维化呈现缺氧状态。研究发现,当PDAC处于缺氧环境时,DNMT3B水平升高使miR-485-3p启动子超甲基化进而降低表达量,减弱miR-485-3p对性别决定区Y框蛋白9(SOX9)和溶质载体家族7成员11(SLC7A11)表达的抑制作用。SOX9可以维持胰腺癌细胞的干细胞样特征进而增强其化疗耐药性。SLC7A11是产生GSH的System XC-的组成部分,其表达水平升高会抑制PDAC细胞铁死亡并增强GEM耐药。基于DNMT3B/miR-485-3p/SLC7A11轴在缺氧条件下对胰腺癌细胞干细胞样维持和GEM耐药机制的进一步研究是改善GEM耐药的有效途径。
GDMCN2是一种用于耐药性胰腺癌治疗的新型药物递送系统,主要由可以诱导肿瘤细胞铁死亡的Fe-MOF(铁基金属有机框架)和共价有机框架组成,并通过抗体实现胰腺癌特异性靶向递送。近年来,研究人员开发了胰腺癌特异性的单克隆抗体NRP2 mAb,并将其与携带有GEM的GEM-DVDMS@Np-MIL-101(Fe)复合物表面连接,增强了肿瘤靶向性,从而使该系统能够快速到达肿瘤部位。声动力治疗利用高能声波产生机械振动,并通过金属有机框架MOF纳米载体提高疗效,增强药物的溶解度、稳定性和浓度。当产生声动力刺激时,该系统会在肿瘤部位释放声敏剂与GEM,并在超声波照射下产生大量ROS,导致线粒体损伤、内质网应激反应和DNA损伤,并降低GPX4水平,触发自噬依赖性铁死亡,从而改善GEM耐药的胰腺癌的治疗效果。
4小结与展望
GEM是晚期胰腺癌的重要化疗药物。但随着治疗阶段的推进,GEM的疗效因耐药性而逐渐减弱。近年来许多研究从PUFA过氧化、铁代谢调控和抗氧化系统方面探讨了诱导铁死亡对GEM疗效的改善。然而,借助其他铁死亡调控机制[例如NAD(P)H/FSP1/CoQ10轴、GCH1/Bp/DHFR轴、鲨烯积累等]诱导胰腺癌细胞铁死亡,进而抑制其GEM耐药的策略仍需深入探索。同时,胰腺癌中铁死亡发生与GEM耐药性之间的分子机制还有待于进一步探究。此外,诱导铁死亡来改善胰腺癌对GEM耐药的研究尚处于基础研究阶段,仍待临床试验验证。转化医学领域如新型材料与天然产物开发利用等研究将为GEM耐药的胰腺癌治疗带来新的突破。GEM与顺铂等多药物联合方案对胰腺癌细胞铁死亡的诱导作用,或许也是解决GEM耐药的思路。
全文下载
https://www.lcgdbzz.org/cn/article/doi/10.12449/JCp50737


