继发性和治疗相关急性髓系白血病:特征重叠,轨迹不同
时间:2025-08-26 12:11:12 热度:37.1℃ 作者:网络
治疗相关急性髓系白血病(tAML)和继发于既往血液疾病的急性髓系白血病(sAML)在生物学特征上具有相似性,包括核型异常和特定基因突变,以及患者相关的风险因素。年龄较大和体力状态较差也会导致预后不良,无论是在缓解率还是总生存期方面。然而,这两种疾病在白血病发展轨迹上也存在显著差异。
《Seminars in Hematology》近日发表综述,回顾了tAML和sAML的临床和生物学特征,强调了它们的共同点和不同特征,并讨论了分类问题。还收集了最新的证据,这些证据涉及从骨髓衰竭综合征、骨髓增生异常综合征(MDS)、骨髓增殖性肿瘤(MPN)和MDS/MPN重叠综合征到sAML的白血病发展轨迹,以及在异基因移植背景下供体细胞引起的白血病。此外还回顾了白血病的遗传和获得性易感性,并讨论了治疗现状和未来方向。

介绍和分类
与de novo AML不同,其可能表现为突然的临床事件,继发急性髓系白血病(sAML)几乎都是复杂发展轨迹的结果——无论是通过骨髓增生异常综合征(MDS)等克隆性造血疾病的逐步进展,还是先前细胞毒性治疗的晚期后果。这种既往病史具有显著的生物学、预后和治疗意义。不断发展的分类系统(国际共识分类和2022年WHO分类第五版)反映了从临床表现到遗传推断的二次AML的转变,更加重视AML的分子基础,而非“回忆性”分类。
髓系肿瘤分类系统的发展显著改变了sAML患者原型:以前主要影响老年人(通常有血液恶性肿瘤病史或因实体瘤接受过化疗/放疗),现在sAML包括所有年龄层的患者,特别是随着遗传易感性继发AML的最近增加,这种AML通常比de novo AML更早发生(DDX41突变白血病除外)。此外,由于均预后不良和治疗耐药,既往有MDS史或因原发肿瘤接受细胞毒性治疗的AML历史上被归为一类,但现在它们越来越多地被视为生物学上不同的亚型,每种亚型都有其独特的遗传和发生特征所塑造的独立治疗路径。
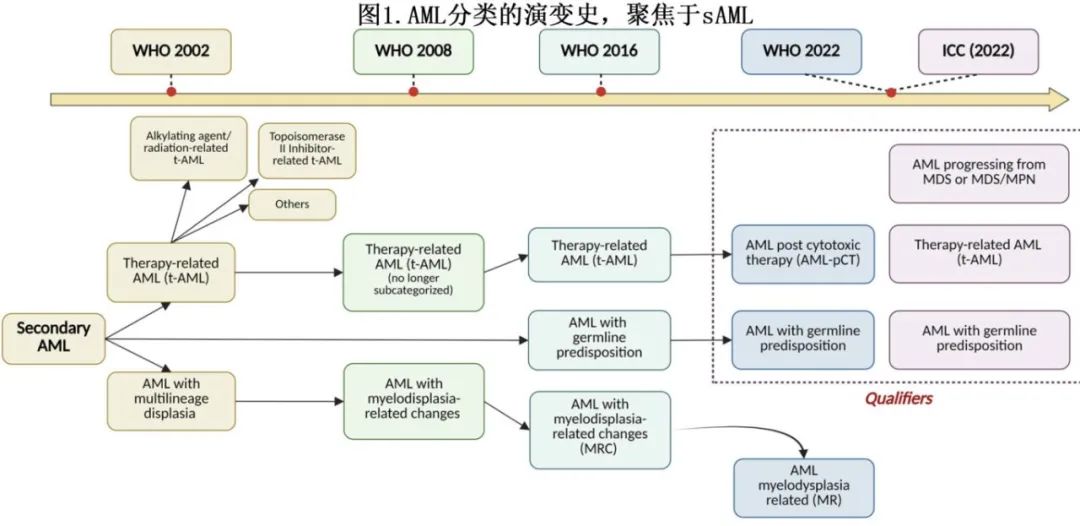
回顾继发性AML分类的发展历程(图1),2002年WHO分类中首次出现了de novo与继发性AML的区别。该版本将sAML包括在内,其诊断标准为骨髓形态学中至少两个髓系谱系的至少50%细胞存在发育异常,或有MDS或MDS/骨髓增殖性肿瘤(MPN)的既往病史,且在AML发病前至少6个月诊断。此外,因先前接受过化疗或放疗治疗无关肿瘤的患者被单独归类为治疗相关AML。治疗相关AML进一步根据致病因素分为与烷化剂/放疗相关的AML(通常在治疗后4-7年发生,常伴有MDS特征)和与拓扑异构酶II抑制剂相关的AML,其特点为潜伏期较短且无先前MDS阶段。
2008年WHO分类修订版取消了治疗相关AML的子分类,并将这些类别以及具有类似特征的MDS病例纳入更广泛的治疗相关髓系肿瘤(t-MN)。此外,随着对白血病克隆细胞遗传特征的深入了解,也促成了与发育异常相关的遗传异常列表的制定,作为新建立的具有骨髓增生异常相关改变的AML(AML-MRC,myelodysplasia-related changes)亚组的诊断标准,取代了多系发育异常(multilineage dysplasia)AML。
2016年,WHO分类第四次修订版对现有类别进行了微笑修改,例如从与发育异常相关的细胞遗传学异常中排除了9号染色体缺失和双等位基因CEBPA突变。此外,首次认可了一类以特定遗传突变(这些突变易导致白血病发展)为特征的髓系肿瘤。
2022年,国际共识分类(ICC)和WHO分类第五版均取消了多系发育异常和平衡的细胞遗传学异常作为诊断标准。根据2022年WHO分类,MDS既往病史、特定细胞遗传学异常的存在以及8个与髓系发育异常相关的(MDR)基因(ASXL1、BCOR、EZH2、SF3B1、SRSF2、STAG2、U2AF1和ZRSR2)的突变定义了具有骨髓增生异常相关AML(AML-MR)。而ICC则提倡一种诊断层次结构,其中分子检测(具有MDR细胞遗传学异常或基因突变的AML,与AML-MR基因相同,另外包括RUNX1)优先于细胞遗传学(具有MDR细胞遗传学异常的AML)。除了常见的异常外,三体8和del(20q)被纳入ICC定义MDS的核型中,而WHO则认可del(11q)和-13/del(13q)。
此外,这两种分类系统还制定了一系列诊断限定词,包括既往接受过细胞毒性治疗(扩展到包括免疫干预和PARP抑制剂)以定义治疗相关AML(根据2022年WHO分类为细胞毒治疗后的AML),以及继发于遗传易感性的AML。ICC不再将从MDS或MDS/MPN进展而来的AML视为一个诊断实体,而是另一个诊断限定词,这与2022年WHO分类不同,后者仍将其视为疾病定义要素。
虽然这两种分类系统在继发AML方面仅存在细微差异,但这些差异可能导致潜在的误读。在临床实践中达成共识具有获益。
易感性
遗传因素
sAML中一个至关重要且常被忽视的方面是遗传易感综合征所致的病例。在这些情况下,白血病的发生并非随机事件,而是由遗传缺陷本身决定的克隆性发展轨迹。白血病易感综合征在所有年龄组中越来越被重视,并挑战了遗传易感性主要与儿童相关的传统观念,即与经典骨髓衰竭综合征相关。历史上,人们认为成人中髓系恶性肿瘤的遗传易感性较为罕见,但大规模基因组测序的进展显著扩展了对与(MDS和急AML相关的遗传变异的理解。2022年WHO分类和ICC现在明确承认遗传易感性的作用,将其作为独立的限定词纳入其中,强化了在特定病例中进行系统遗传评估的需求。随着遗传检测更加普及,识别成人AML患者的遗传贡献对于细化风险分层、指导治疗决策和完善家族咨询至关重要。在异基因造血干细胞移植的背景下,识别潜在的遗传易感性具有重要的治疗意义,因为相关供者可能也携带易感变异。此外,了解遗传易感性与白血病发生之间的分子机制可能揭示新的治疗靶点并改善早期检测策略。展望未来,将遗传检测纳入特定MDS/AML患者的常规临床实践,特别是那些早发性疾病、原因不明的血细胞减少或有家族史的患者,对于改善患者预后和推进髓系恶性肿瘤的个性化医疗至关重要。
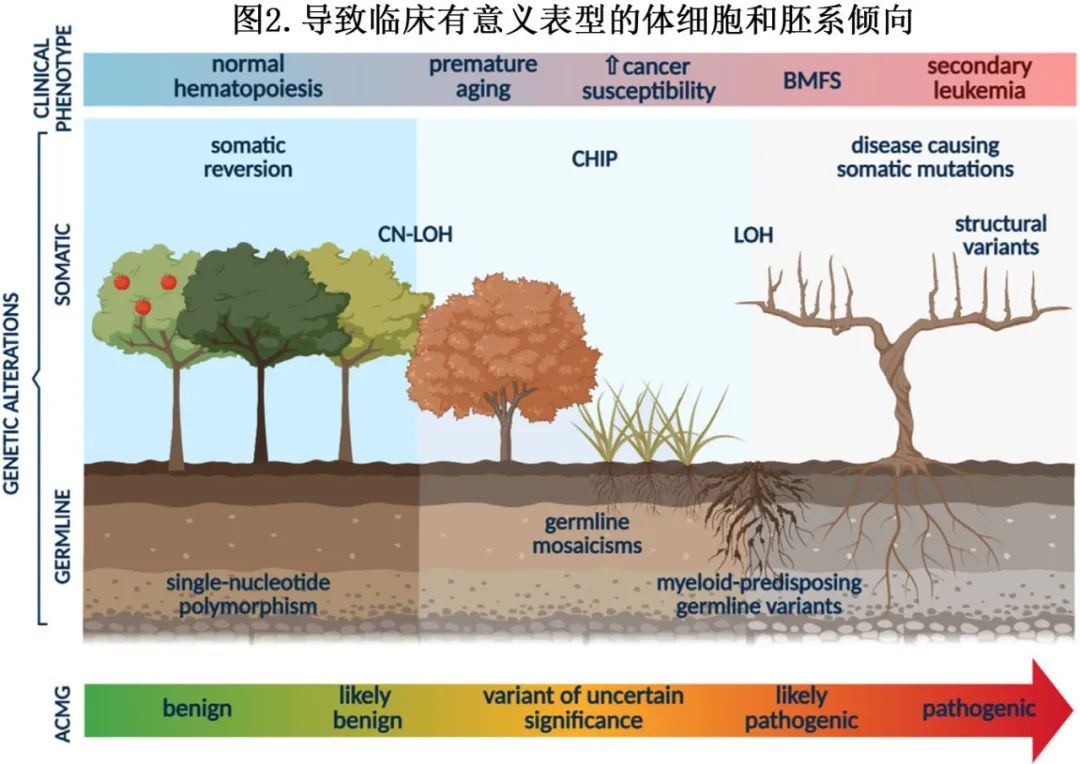
识别遗传易感变异需要与遗传学家密切合作,以操控复杂的遗传改变特征(图2)。一些常规分析用于克隆筛查和疾病监测的变异可能具有遗传或体细胞起源,因此必须通过额外的造血外组织确认其遗传性质。理想情况下,遗传变异在杂合子时的变异等位基因频率(VAF)约为0.5,纯合子时为1.0。在血液来源组织(骨髓或外周血)的NGS分析中,VAF≥0.3可能提示遗传起源(疑似杂合子)。虽然体细胞变异的VAF会随疾病状态波动,但遗传变异随时间保持相对稳定的VAF。然而也存在解释挑战,因为VAF可能受遗传嵌合体、细胞遗传学重排或反向突变等因素影响。
涉及遗传变异的一个主要问题是确定其病理意义。美国医学遗传学学院(ACMG)指南为变异解释提供了基础框架,但它们缺乏对髓系易感基因的特异性,特别是那些尚未被验证为单基因疾病的基因,但其与衰老和基因组不稳定性相关——这些因素可能导致癌症易感性。需要进一步完善分类标准,以提高对髓系恶性肿瘤中遗传变异解释的准确性。
遗传背景与体细胞景观密切相关,诊断技术用于在分子和细胞遗传学水平检测克隆事件。NGS特别有价值,可用于识别结构性变异、无拷贝数变化的基因组改变、小插入和缺失(indels)以及单核苷酸变异。染色体显带技术和荧光原位杂交(FISH)对于检测大片段拷贝数变异(CNVs)、易位、倒位和非整倍体至关重要。此外,单核苷酸多态性阵列(SNP-A)已引入临床实践,用于检测宪法嵌合体、大片段CNVs和杂合性丢失区域(LOH),包括复制中性LOH(CN-LOH,也称为单亲同源二倍体—UPD)。
不同的克隆途径似乎与潜在的易感性紧密相关,从而定义了继发性白血病的发生轨迹。在经典骨髓衰竭综合征中,这一过程通常以一系列连续的克隆事件为特征。在严重的先天性中性粒细胞减少症(SCN)中,获得CSF3R截短突变和RUNX1改变定义通向AML的主导克隆途径。ELANE突变的SCN亚型最为常见,但涉及同一途径的白血病进展也在携带遗传性WAS、HAX1、G6PT1变异的病例中有所描述。同样,在Schwachman-Diamond综合征(SDS)和ERCC6L2相关疾病中,白血病转化经常涉及p53通路,需要双等位基因TP53突变,并反映出DNA损伤响应中的共同脆弱性。在范可尼贫血(FA)中,p53通路的间接损害也被描述为白血病转化的前体。FA患者可能获得1q染色体 gains 导致MDM4扩增和p53通路下调。在SAMD9/SAMD9L综合征和GATA2缺乏等MDS/AML易感条件下,单体7 呈现为早期复发事件。在SAMD9/SAMD9L综合征中,这通常代表一种选择性机制,以消除有害的遗传等位基因。
与该模型不同,DDX41和CEBPA突变AML遵循经典的“二次打击”模型。虽然单一的遗传变异似乎现在不足以促进血液学表型,但在细胞适应性约束的影响下,同一基因中发生的体细胞突变(通常影响第二个等位基因)是完全白血病转化所必需的。
这些模式强调了遗传背景如何塑造白血病发生的进化限制和机会,以及在具有遗传易感综合征的个体中个性化监测和根据风险调整管理的需求。
获得性
AA和PNH
再生障碍性贫血(AA)和阵发性夜间血红蛋白尿(PNH)是主要依赖T细胞介导自身免疫的骨髓衰竭综合征。约10%的AA患者在随访期间发展为PNH,因此促使人们假设在自身炎症性自身免疫背景下PNH克隆具有生存优势。这两种疾病都伴有相当比例的继发性髓系恶性肿瘤(sMN),在AA中占15%至20%,在PNH中占2%至6%,且发生在初始诊断后10年,是最重要的长期严重并发症。
自Lane等首次报告以来,许多学者试图描绘AA患者克隆造血(CH)的特征,报道的突变频率介于5%-70%。一项最大的国际研究纳入439名AA患者和668份血液样本,进行了全面的NGS髓系筛查(82名患者在多个时间点进行了检测)。作者在156名患者中检测到249个体细胞突变(占研究人群的36%),最常见的是影响BCOR和BCORL1(9.3%的患者)、PIGA(7.5%)、DNMT3A(8.4%)和ASXL1(6.2%)。值得注意的是,除了PIGA和BCOR/BCORL1外,多数突变具有与年龄相关的特征。此外,PIGA、BCOR和BCORL1突变与治疗反应改善、总生存期和无进展生存期相关。通过纵向筛查显示,CH在大多数情况下起源于诊断时存在的一小部分克隆。克隆轨迹呈异质性:部分克隆多年来保持稳定,另一些则占据血液部位,促进亚克隆的出现,且先于sMN的发展。
为了评估AA/PNH中克隆演变相关的临床和分子特征,学者回顾性分析了1008名患者。除了PIGA突变外,最常突变的基因是DNMT3A、ASXL1和BCOR/L1,sMN的10年累积发病率为11.6%。进展风险最高的患者是患有严重AA或对首次或挽救治疗未达到完全反应的患者。与匹配的de-novo病例相比,AA/PNH后的MDS呈现更高的R-IPSS评分(54% vs 37%)、更频繁的7号染色体异常(53% vs 11%)以及ASXL1(24% vs 12%)或RUNX1(21% vs 8%)突变。
考虑到有证据表明特定人类白细胞抗原(HLA)类或单倍型表达与AA/PNH的发病机制相关,多个研究小组调查了HLA与白血病进展之间的相关性。Hosokawa等分析了633名AA患者,发现有HLA类I类缺失(HLA−)白细胞的患者(127例,20%)中无人进展为sMN,而这一并发症在没有HLA−白细胞的患者中10年发生率为5.8%。Zaimoku等分析了544名AA患者,显示携带HLA−白细胞的患者比例相似(22%),然而与之前的结果相反,该特征与高危克隆演变相关。这一关联后来未被同一研究小组确认。最近,Pagliuca等报告了HLA异常与sMN发病时较低的突变负担之间的相关性,但在sMN进展方面没有差异。
、
总而言之,AA/PNH后的MN进展仍然是一个未满足的医疗需求;作为预测生物标志物的白细胞上HLA类I类的缺失以及包括PIGA、BCOR和BCORL1在内的特定突变的作用需要进一步验证。
LGLL
大颗粒淋巴细胞白血病(LGLL)与sMN之间的关系尚不明确。LGLL是一种罕见疾病,其特征是具有细胞毒性的克隆性大颗粒淋巴细胞(LGLs)的慢性异常增殖,其病因病理学仍存在争议,因为其临床表现范围广泛(从惰性肿瘤到危及生命的情况)。LGLL还与自身免疫性疾病、AA和纯红细胞再生障碍症有重叠特征。
此外,LGL克隆在髓系肿瘤中也有发现。2013年,Jerez等人描述了24例同时患有MDS和LGLL的病例(其中9例存在STAT3突变),并在343例已知MDS患者中检测了STAT3突变的LGLL克隆,发现另外9例(2.6%)。同样,Ai等人分析了721例MDS患者,发现10例(1.4%)存在LGL克隆。相比之下,2020年,Moffitt癌症中心的研究人员分析了1177例MDS患者,通过流式细胞术发现27%的病例存在LGL克隆扩增。在治疗反应(包括来那度胺、去甲基化药物、促红细胞生成剂或免疫抑制治疗)和生存方面,LGL+和LGL- MDS之间没有观察到差异。
Durrani等人进行了一项反向研究:分析了240例LGLL患者,发现13例MDS(5.4%)。LGLL/MDS和LGLL队列中STAT3/STAT5突变率、LGL计数、TCR重排和诊断时的平均年龄相似。值得注意的是,LGL/MDS患者比LGLL患者更常出现血小板减少症(P=0.014),而后者的单系血细胞减少症更为常见。在分析基因组特征时,MDS/LGL、MDS和LGL患者的个体突变分布总体相似,但剪接体突变在MDS中更为常见,而DNMT3A、TP53和NPM1突变在MDS/LGL中更为常见。
从上述证据来看,无法确定LGL扩增与MDS克隆出现之间的时间顺序,也无法确定该克隆的性质(继发性还是原发性MDS)。Kawashima等人最近的研究提供了相关线索:分析了349例LGLL,发现8%的病例与MN重叠,另外19%存在CH,其中TET2(23%)和DNMT3A(14%)是最常见的驱动因子。与1443例CHIP个体队列相比,LGL患者中DNMT3A突变的频率较低,而SRSF2、U2AF1、KRAS、MPL、RUNX1、KIT、IDH1和NPM1突变的频率较高。此外,CH+ LGL患者与CH- LGL患者相比,MN的发生率显著更高(5年分别为10%和2%)。这些发现表明,LGL与特定类型的CH相关,这些CH由具有高致癌潜力的突变基因驱动,这也得到了后续MN高发病率的证实。
细胞毒治疗
细胞毒性治疗的类型和时间是发生治疗相关 AML 的关键步骤。事实上,正如2002年和2008年 WHO 分类所强调的,不同化合物或细胞毒性治疗的作用机制转化为不同的疾病表型和原始治疗的潜伏期。最常与治疗相关 AML 相关的治疗见表1。

继发性白血病的机制
继发于MDS的AML
MDS和sAML之间的界限常常不清晰,因为它们具有重叠的生物学特征,并且是髓系疾病谱系中的一个连续体。最近,Kewan等人在3598例MDS/sAML的大样本研究中利用无监督聚类方法,对这两种疾病的区别提出了挑战,并识别出14个由分子和细胞遗传学特征定义的聚类(并非由诊断或原始细胞比例定义)。这些发现反映了基于分子的分层优于基于临床的分类。然而,尽管存在显著的重叠,仍可以在MDS/sAML的连续体中区分早期和晚期事件。特别是,表观遗传修饰基因(如DNMT3A、TET2)、TP53和剪接体基因(如SRSF2、SF3B1、ZRSR2、U2AF1)的突变在MDS早期出现,而RAS通路(如NRAS、KRAS)、受体基因(如FLT3)和转录因子(如RUNX1、GATA2、CEBPA)的突变则在白血病发生过程中较晚出现。尽管有多个报告描绘了突变发生的发育/层次顺序,但突变的机制和组合在恶性克隆中可能是异质的。事实上,某些突变更频繁地共同发生(例如SETBP1与c-CBL和ASXL1,或SF3B1与TET2),而其他突变则相互排斥,如IDH1/2与TET2或多个剪接体基因,可能遵循通路冗余的原则。此外,这种突变获得模式可以在同一位患者中同时发生,由不同的克隆引发,这些克隆可能或可能不存在层次关系,从一个创始克隆开始,获得额外的亚克隆。最近的研究表明亚克隆在MDS进展中起主导作用,其从创始克隆的分支可以是顺序的或平行的,多个亚克隆可能从同一个创始克隆中出现。Makishima等人通过分析顺序样本观察到,一个亚克隆可以渗出一个或多个预先存在的克隆并成为主要克隆。
MDS的国际工作组(iWG)提出了MDS向sAML进展的预测,经过多年发展,开发了多种风险模型。特别是最近发布的分子IPSS(M-IPSS)保留了IPSS-R中识别的细胞遗传学风险等级,保留了骨髓原始细胞比例、血小板减少和贫血作为临床变量(去除中性粒细胞减少),并纳入16个预后基因的17个二元特征和15个基因残余组的特征。
继发于MPN的AML
费城染色体阴性MPN是一组具有不同临床特征和遗传驱动因素的克隆性疾病,其发展为继发性AML的风险在10年内各不相同。在原发性骨髓纤维化(PMF)中这一风险为10-20%,在真性红细胞增多症(PV)中为2%-4%,在原发性血小板增多症(ET)中为1%-2%。与继发性演变相关的临床特征包括年龄较大、白细胞增多、循环原始细胞存在以及血小板减少和/或贫血恶化。对于骨髓纤维化(MF),特定风险因素包括血清IL-8和C反应肽水平升高,而既往有血栓形成史与ET中的sAML相关。暴露于pipobroman、白消安和32P与sAML风险增加有关,而羟基脲的相关证据则存在矛盾。
在生物学层面上,不同MPN表型的核型异常发生率差异很大(ET <5%,PV <20%,MF 30%-60%)。复杂核型、i(17q)、-5/5q-、-7/7q-、+8、12p-、inv(3)和11q23重排等与继发性演变更常见相关,并已被纳入用于预测PMF演变的DIPPS plus评分系统。然而,多变量分析仅确定血小板计数低于100×10⁹/L和不良核型(而非DIPSS plus评分)为白血病无进展生存的独立预测因素。
分子研究进一步阐明了PMF中的白血病风险,促成了新的评分系统(如MIPSS、MIPSS70、MIPSS70 plus、MIPSS70 plus 2.0、GIPSS),这些系统包括定义疾病遗传多样性的分子变量(如ASXL1、EZH2、IDH1/2、SRSF2、U2AF1突变)。最近多个研究团队报告称,NRAS、KRAS、c-CBL、RUNX1和TP53突变对总体生存期、无事件生存期以及治疗耐药具有预后影响。法国研究团队报告了TP53突变与白血病转化之间的关联。Coltro等人的研究显示,RAS基因突变可预测JAK抑制剂耐药和更高的白血病演变率。Mylonas等人也表明,在PMF患者队列中,JAK抑制剂的使用与随时间推移复杂的克隆轨迹发展以及不相关克隆的出现相关。在移植患者中,CBL突变(涉及RAS通路并与慢性髓系表型相关)对总生存期和非复发死亡率有影响。2024年,Loscocco等人分析了363名患者的大型队列,除TP53外,未确认这些突变在MIPSS70/MIPSS70plus/ MIPSS70plus2.0之外的独立价值。需要进一步的研究来确认这些结果并评估TP53的作用。有趣的是,在JAK2突变MPN继发性白血病发生过程中,JAK2突变克隆通常丢失,而包括CBL、TET2和TP53在内的其他突变则获得。这一现象可以用JAK2阴性祖克隆的扩展和转化来解释。
单细胞研究揭示了MPN后发生AML的复杂克隆轨迹,挑战了基于原始MPN驱动突变的多步骤过程的经典假设。Lundberg等人显示,DNMT3A和TET2突变可能先于JAK2突变或作为独立克隆共存。反之,ASXL1、TP53以及在某些情况下的TET2突变,也可能代表白血病发生过程中的晚期事件,表明相同基因突变在继发性AML发展中可能具有不同的作用。
继发于MDS/MPN的sAML
骨髓增生异常/骨髓增殖性重叠综合征(MDS/MPN)是一组具有异质性的髓系肿瘤。2022年, WHO第五版和ICC更新并扩展了MDS/MPN组,现在包括慢性粒单核细胞白血病(CMML)、MDS/MPN伴中性粒细胞增多或非典型慢性髓系白血病(aCML)、MDS/MPN伴SF3B1突变和血小板增多(MDS/MPN-ST)以及MDS/MPN非特指型(NOS,不满足明确定义类别的标准)。MDS/MPN亚型在临床表型、生物学特征和预后方面存在显著差异。例如,CMML富集与粒单核细胞扩增相关的突变(如TET2、ASXL1、RAS通路基因)和SRSF2;aCML则富集ASXL1、SETBP1和TET2基因突变以及三体8核型异常;MDS/MPN-ST通常具有SF3B1和JAK2基因突变,且核型通常正常。MDS/MPN NOS的基因组谱高度异质。Palomo等对106例患者进行的最大规模研究显示,ASXL1、TET2、JAK2、EZH2和SRSF2是最常突变的基因。根据分子谱,作者确定了4种MDS/MPN NOS亚型:CMML样(17%的患者)、aCML样(33%)、MDS/MPN伴环形铁粒幼红细胞和血小板增多(MDS/MPN-RS-T;11%)和TP53(14%)。其余患者未显示独特的基因特征,但富集U2AF1、JAK2和ASXL1突变。
这些亚型的生物学异质性也反映出继发性白血病发生风险和总体预后的差异。多个风险评分和预后研究涉及CMML预后,包括的变量有CBC参数、原始细胞计数、细胞遗传学异常和高危基因突变。所有这些评分在预测CMML患者的LFS和OS方面表现良好。最近制定的BLAST评分整合了临床变量和遗传信息,其性能与分子CMML特异性预后评分系统(CMML)和IPSS-M相当。原始细胞转化的独立风险因素包括DNMT3A和ASXL1基因突变、PHF6基因野生型、白细胞≥13×10⁹/L以及循环≥2%或骨髓原始细胞≥10%。MD安德森中心最近的一项研究确定了189例由CMML演变而来的AML患者的基因组和临床特征。作者确定了3种主要的白血病发展轨迹:单核细胞AML(53%),其特征为单核细胞和早单核细胞扩增以及SRSF2、TET2和RAS突变;髓系AML(43%),其特征为未成熟髓系原始细胞扩增和CEBPA突变;红细胞AML,其特征为复杂核型和TP53突变。
其他亚型的白血病风险和总体预后方面研究较少。aCML的中位OS在10到16个月之间,1年白血病转化率在2%到12.5%之间。MDS/MPN-ST的预后最好(在Palomo等分析的71例患者队列中,中位OS未达到,中位随访时间为48个月)。Mangaonkar等分析了158例患者,在62个月的中位随访中观察到6年的中位OS,且sAML风险较低(Palomo等报告为9%,Mangaonkar等报告为4%)。MDS/MPN NOS的预后和白血病风险尚不明确:MD安德森癌症中心的85例研究报道中位OS为12个月,而梅奥诊所-莫菲特癌症中心联合研究包括135例患者,在61个月的中位随访中,OS和LFS分别为26个月和24个月。有趣的是,Palomo等基于分子的MDS/MPN NOS分层在CMML样、aCML样、MDS/MPN-RS-T样和TP53样中具有预后意义,接近相应MDS/MPN亚型的预后,其中TP53组患者的预后最差。
治疗相关
t-MN是原发肿瘤和自身免疫疾病细胞毒治疗最严重的长期后果之一。这些疾病预后不良,对当前治疗反应差,约占新诊断AML/MDS)的10%至20%。其不良预后可能反映出复杂核型(约48%)和TP53突变(30%-47%)的高发生率,这两种异常在约80%的病例中同时存在。
尽管在过去的20年中,多个研究小组致力于确定t-MN发展中的主要风险因素,这些因素可分为患者特异性(年龄、既往癌症、自身免疫疾病和环境暴露)、遗传性(多态性和遗传突变)和获得性(CHIP和慢性炎症),但数据解释依然复杂。识别t-MN发展中的关键风险因素的主要困难在于研究队列规模小(罕见疾病)、原发肿瘤治疗方案不同以及新药替代旧药。
最近,随着深度测序技术的应用,发现携带CHIP的个体中t-MN发生率更高。
虽然最初发现CHIP时认为其通常为良性,但现在认为细胞毒治疗(如化疗和/或放疗)可能创造一个选择性环境,促进预先存在的耐药克隆扩增,可能导致t-MN的发展,无论是否获得额外的遗传改变。这种现象现被称为治疗相关克隆性造血。
在CHIP/年龄相关克隆性造血(ARCH)发现后的几个月,Wong等通过NGS筛选22个t-MN的基因组,发现7个TP53突变携带者(32%)。通过高通量NGS和数字PCR(ddPCR)进行的回顾性分析,在7名患者中的4名(57%)原发肿瘤诊断时的配对DNA样本中发现了相同突变,变异等位基因频率低(0.003%-0.7%),首次揭示了TP53突变在治疗相关急性髓系白血病起源和进化中的作用。
后续研究,包括本文作者对14名有血液恶性肿瘤史的t-MN患者的研究,确认并扩展了这一模型。使用超深度NGS,作者发现了两种不同的进化模式:3名患者在任何治疗前即可检测到体细胞突变,证实诊断时已存在白血病克隆;而另外5名患者的突变仅在细胞毒暴露后出现,支持治疗诱导的起源或选择。值得注意的是,在已识别的基因中,TP53仍然处于中心地位,但ASXL1、IDH1和SRSF2的额外突变也有助于克隆演变。此外,在一个典型病例中,IDH1突变在t-MN发生前9年以稳定VAF(35%)存在,表明存在休眠的白血病前期阶段。后续获得的SRSF2突变可能触发白血病进展,表明存在序贯多步骤的发病机制。
现在很明显,克隆造血不仅有助于t-MN的发展,而且这种贡献直接受患者接受的具体治疗的影响。因此,研究人员现在正专注于研究具有相同原发肿瘤和接受同质治疗的患者队列,旨在识别赋予特定CH突变选择优势的治疗暴露。在这方面,Sperling等在一个416例t-MN患者队列中发现复杂核型与铂类药物暴露之间存在直接相关性;涉及5号和7号染色体的染色体异常与烷化剂暴露相关;TP53突变与沙利度胺类似物和蛋白酶体抑制剂暴露相关,特别是在多发性骨髓瘤病史的患者中。有趣的是,在继发性白血病发生背景下出现的核型异常呈现特定的突变情况:Halik等在519例7号染色体畸变的 AML 患者人群中观察到 SRSF2 和 U2AF1 突变在 sAML 中富集。
值得注意的是,通过对经过CRISPR-Cas9改造的小鼠的造血干细胞和祖细胞(HSPCs)进行长期体外竞争实验,发现来那度胺(而非泊马度胺)选择性地促进TP53突变HSPCs的克隆扩张。相比之下,具有PPM1D、TET2或DNMT3A突变的细胞在相同的治疗压力下并未表现出这种正向选择。
最近,作者研究小组报告称,在接受化疗(免疫治疗)后发展为t-MN的慢性淋巴细胞白血病(CLL)患者中,CHIP的发生率很高,最常用的治疗方案是FCR方案(氟达拉滨、环磷酰胺和利妥昔单抗)。在13例进展为t-MN的CLL患者中,在10例(77%)中发现了30种致病性或可能致病性的CHIP变异,而在对照组的285例CLL患者中仅为12%(34例)。值得注意的是,对诊断样本的回顾性分析显示,在62.5%的t-MN病例中,这些突变在初始CLL诊断时即可检测到。
Yan等人强调了在霍奇金淋巴瘤自体移植中CHIP作为t-MN风险因素的作用。通过分析一个由321例患者(中位年龄为34岁)组成的回顾性队列中通过单采获得的HSPCs,CHIP发生率为14.3%(46/321),主要由DNMT3A(n=25)、PPM1D(n=7)、TET2(n=7)和TP53(n=5)突变引起。与文献一致,携带高危突变(PPM1D和/或TP53)的患者在非复发死亡率和t-MN累积发生率方面预后极差。
识别CHIP作为t-MNs的前体对肿瘤患者的风险分层和临床管理具有重要意义。早期识别CHIP携带者可能实现更有针对性的监测和潜在干预措施,以降低t-MN发展的风险。
特殊场景
继发于CAR-T的AML
嵌合抗原受体T细胞(CAR-T)是一种自体工程化构建物,具有抗原识别的细胞外结构域、跨膜结构域和由共刺激结构域和分化簇3(CD3 ζ信号尾)组成的细胞内结构域。CAR-T疗法在淋巴系统恶性肿瘤中取得了突破,迅速成为急性淋巴细胞白血病(ALL)和B细胞淋巴瘤的挽救治疗策略,而其在髓系恶性肿瘤中的应用仍不佳。
最近,越来越多的证据报告了CAR-T输注后sMN和T细胞淋巴瘤的发生。曾报告一例33岁男性患者,患有难治性大B细胞淋巴瘤,CAR-T治疗是其第七次治疗方案。在CAR-T治疗时,骨髓检查未显示任何发育异常迹象,核型正常。CAR-T治疗前进行的NGS发现了CBL、DNMT3A和JAK3突变。CAR-T输注后六个月,患者诊断为t-AML,与DNMT3A克隆倍增以及RUNX1和FLT3突变克隆的出现相关。
Alkhateeb等人分析了梅奥诊所的189名接受CAR-T治疗的患者,报告了5.3%的t-MN发生率。Cordeiro等人分析了86名接受CD19 CAR-T治疗的患者,报告了4例MDS病例(4.6%),其中2名患者在接受CAR-T治疗前已有细胞遗传学异常。
2024年,Hamilton等人报告了斯坦福大学治疗的724名CAR-T患者的长期随访结果。中位随访15个月,观察到25例继发恶性肿瘤,其中14例为血液系统恶性肿瘤。其中,13例为髓系恶性肿瘤(1.8%;10例MDS,2例AML,1例MDS/AML)和1例T细胞白血病。有趣的是,作者发现T细胞白血病和原始大B细胞淋巴瘤中存在共同的由DNMT3A和TET2驱动的克隆造血。
这些报告对于CAR-T治疗与sMN之间的整体相关性的解释并不容易。需要记住的是,所有患者在接受CAR-T治疗前都接受了多线强化疗,可能引入了潜在偏差。事实上,t-MN的发生率与接受多线强化治疗的非霍奇金淋巴瘤(NHL)患者中报告的发生率并没有显著差异。Calip等人分析了SEER数据库中18245名接受化疗和/或免疫治疗的老年NHL患者,在中位随访3.5年后观察到3.7%的继发MDS/AML。由国际血液和骨髓移植研究中心(CIBMTR)也报告类似结果,该中心分析了9028名接受自体干细胞移植的NHL患者,其中335例在移植后诊断为t-MDS/AML(3.7%)。
供者来源的AML
在异基因造血干细胞移植(allo-HSCT)后的MN复发中,很少发现供者来源的白血病(donor derived leukemia,DDL)。
从1971年首次报道的病例开始,有几篇文章报道了供者来源急性髓系白血病(DDL-AML)病例,其性质通过细胞遗传学或基于PCR的供体嵌合体来确定。特别是,欧洲血液和骨髓移植集团(EBMT)的一项调查,包括91个中心,覆盖10489例造血干细胞移植(HSCT),报告了14例(0.1%)供者来源白血病病例(7例AML,3例ALL,1例慢性粒细胞白血病)。日本的一个大型联合研究收集了43788例患者,这些患者接受了异基因(n=18874)或自体HCT(n=24914),用于治疗非髓系恶性肿瘤或非恶性疾病。其中352例患者发生MN(304例为自体HSCT后,48例为异基因HSCT后)。值得注意的是,异基因HSCT后的MN患者中有一半(24例患者)为供者来源,且比真正的治疗相关MN出现得晚(中位时间:26个月 vs 6个月)。2019年,另一份来自EBMT的报告确定了DDL的预估发病率为每10万例移植中有80.5例。
最近,更精细的分子分析使得在移植后能够检测和描述克隆轨迹。特别是,有研究分析了从造血干细胞移植到DDL的白血病发生过程(表2)。
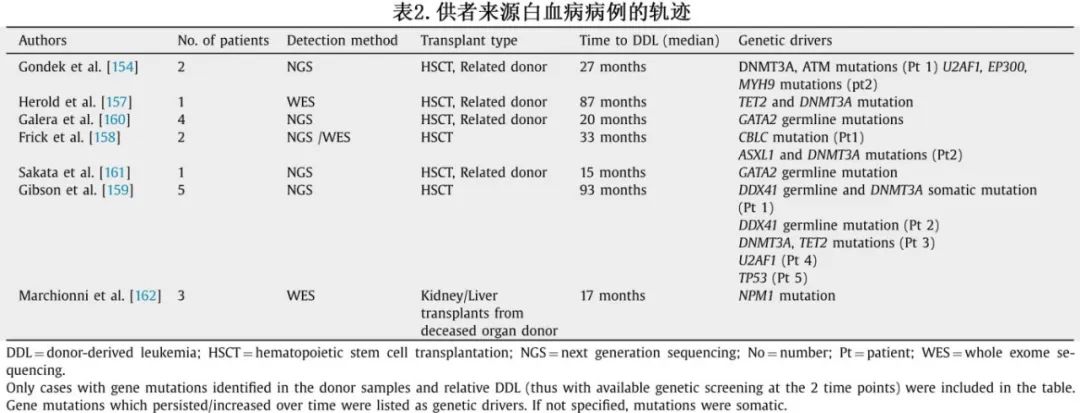
Gondek等人分析了61例老年(>60岁)供者的HSCT,其中两名受者发展为DDL。第一名患者在移植后36个月发展为AML,第二名患者在移植后18个月;通过NGS比较供者样本和DDL的遗传特征,研究人员发现第一名患者中DNMT3A和ATM突变的变异等位基因频率(VAF)增加了3倍以上,第二名患者存在U2AF1、EP300和MYH9突变。Herold等人报告了一名男性患者接受其HLA匹配的姐妹的HSCT。7多年后该患者发展为AML,此后供者本人也发展为AML。供者样本和DDL的比较分析显示存在DNMT3A和两个TET2突变。与CHIP和t-MN中的克隆轨迹观察一致,DNMT3A克隆的VAF保持稳定,而在TET2突变中,一个保持稳定、另一个在白血病发作前稳步增加。
Frick等人和Gibson等人研究了供者CHIP对HSCT结局的影响。Frick等人报告了与慢性移植物抗宿主病(GVHD)的相关性以及复发/进展率的降低,Gibson在描述DNMT3A-CHIP时也描述了同样的发现两位作者报告了受者中CHIP克隆的高嵌合率,并描述了DDL的异质性克隆轨迹,其遗传驱动因子列在表2中。
少数作者报告了作为DDL主要基因驱动因子的生殖系突变的病例,包括GATA2和DDX41,强调了在选择亲缘供者时需要进行仔细的家族分析。
最后,Marchionni等人描述了一个有趣的病例,三名器官受者(一名患者接受肝脏移植,另外两名患者接受肾脏移植)来自同一位去世的供者。所有受者在中位潜伏期17个月后均发展为NPM1突变AML。在供者中也发现了相同的突变。
值得注意的是,也有少数报道在供者中未检测到突变的DDL病例,从而导致了以下假设:(1)靶向测序未涵盖驱动基因突变;(2)考虑到某些病例中较长的潜伏期,可能是供者细胞在环境中发生de novo白血病。
生物特征与临床展望
尽管发展轨迹不同,但sAML和tAML具有相似的生物特征,例如高危基因突变和不良风险核型(图3)。
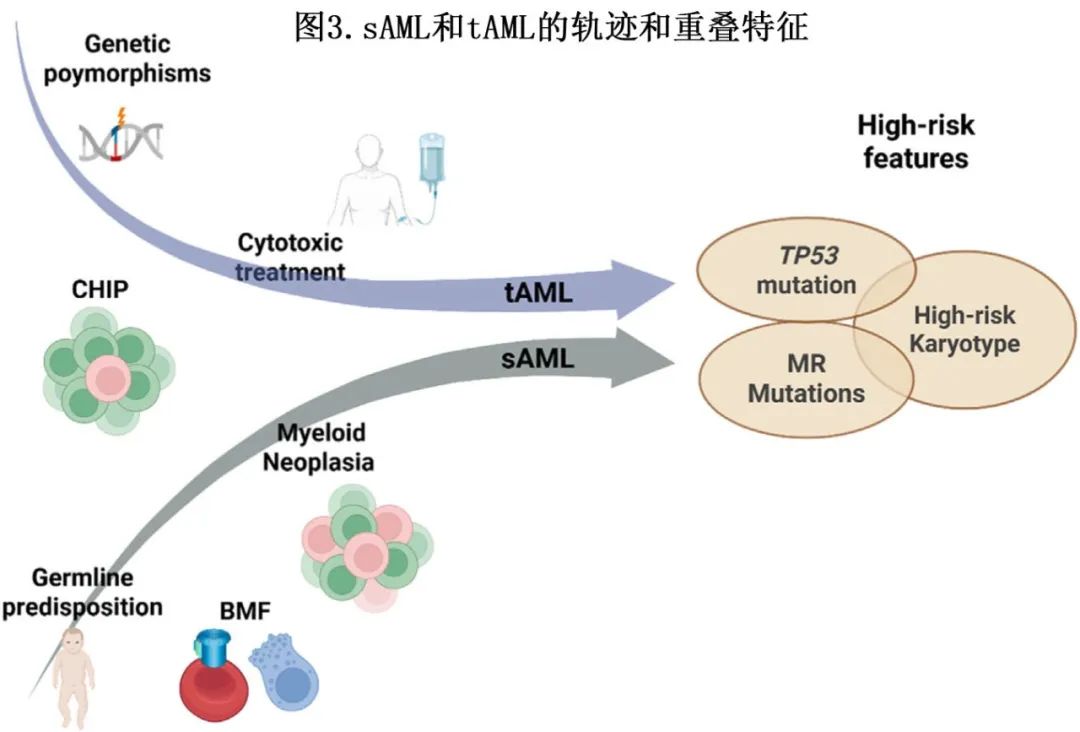
尽管文献中没有直接比较先前髓系疾病后发生的sAML和tAML生物特征的研究,但仍可识别出这两种疾病之间的一些偏好异常:tAML更易出现复杂核型(40%-50% vs sAML的25%)、del(7)(40-50% vs 30%)和TP53突变(25%-40% vs sAML的15%-20%),而sAML则更易出现影响ASXL1(30% vs tAML的3%-15%)、TET2(25% vs tAML的5-15%)和SRSF2(20%-25% vs tAML的10%)基因的突变。由MDS引发的sAML在特征上介于继发于MPN的sAML和tAML之间,具有高发的复杂核型、del(5)和del(7)(25%-40%),以及TP53、SRSF2、SF3B1、EZH2、TET2(20%-35%)基因突变。
从临床角度来看,经典的3+7阿糖胞苷-柔红霉素诱导方案在sAML和tAML中的疗效非常有限,完全缓解(CR)率在20%到50%之间,中位总生存期(OS)为5到10个月。由Lancet等人主导的III期试验测试了一种双药脂质体封装的阿糖胞苷和柔红霉素(固定5:1协同摩尔比),即CPX-351,该试验在sAML治疗中具有重大变革意义。该试验以1:1的比例随机分配了309名高危AML/sAML患者接受CPX-351或3+7方案治疗。CPX-351可改善OS(1年OS为41.5% vs 3+7组的27.6%)和无事件生存期(EFS)。一项事后分析显示,无论是否接受过去甲基化药物(HMA)治疗,tAML、有先前MDS和CMML史的AML患者均显著获益于CPX-351治疗。因此,CPX-351成为fit新诊断sAML和tAML的标准治疗方案。
对于不适合强化疗的unfit/老年患者,可从HMA与BCL2抑制剂维奈克拉(VEN)联合治疗中获益。这一标准治疗方案由Di Nardo等人在2020年通过VIALE-A试验确立,该试验将老年患者(>75岁)随机分配到接受阿扎胞苷(AZA)-VEN或AZA-安慰剂治疗组。AZA-VEN组实现更高的复合CR(CR或伴有不完全血液学恢复的CR;66.4% vs AZA-安慰剂组的28.3%)和改善的OS(14.7个月 vs AZA-安慰剂组的9.6个月)。在sAML亚组,AZA-VEN的优势同样显著(16.4个月 vs AZA-安慰剂组的10.6个月)。长期随访分析进一步证实了这些发现。此外,在AZA-VEN组的多变量分析中,OS与年龄、性别、ECOG体能状态或AML状态(原发vs继发)无显著差异。
根据ELN2024分层,继发性突变通常不具有不良预后影响(TP53突变除外)。HSCT仍是唯一的治愈策略,但由于高龄、体能状态差和难以获得临床反应,只有少数患者适合这种治疗策略。
少数研究专门针对MPN加速期(A-MPN)和由MPN引发的sAML。2018年,McNamara等人报告了108例A/BP-MPN患者的病例系列(44例接受强化治疗,27例接受非强化治疗,51例接受最佳支持治疗)。作者发现,强化治疗与非强化治疗在OS方面无差异,且强化治疗的获益仅限于能够接受移植的患者。此外,TP53是唯一与较短OS相关的突变。Tefferi等人描述了410例A-MPN患者。中位OS非常糟糕(3.6个月),HSCT、复合CR但无HSCT、高危核型和血小板计数是独立风险因素。西班牙PETHEMA小组的一份报告描述了372例在MPN或MDS/MPN后诊断为AML的患者(140例接受强化疗,118例接受非强化策略,114例接受最佳支持治疗),总体中位OS为4.8个月。强化疗的CR率(43%)高于非强化疗(12%)或HMA(8.5%),且与接受非强化疗的患者相比,接受强化疗或HMA的患者OS更长(中位数:8.5个月 vs 8.6个月 vs 4.2个月)。根据先前疾病亚型,CR率或OS无差异。最近,Davidson等人对138例A/BP-MPN患者进行了回顾性分析,这些患者接受强化(N=81)和非强化(N=57)策略治疗。接受强化治疗的患者更常实现复合CR(29.6% vs 非强化组的7.4%)。TP53和RAS通路突变与强化治疗患者的治疗反应较差相关,而较差的体能状态与强化和非强化治疗组的治疗反应均较差相关。
总结
基因组学的进步使我们能够更准确地界定治疗相关和继发性白血病在形成过程中的共性与差异。如今,AML的分类也反映了这一点,明确区分了这两种疾病亚型。尽管它们的恶性发展轨迹差异显著,但最终却呈现出相似的临床特征和糟糕的预后。值得注意的是,基因组筛查的广泛应用使我们能够更精准地对这些白血病的突变图谱和核型异常进行分层,从而识别出 sAML 与 tAML 更具特异性的标志物。然而,尽管我们对白血病的发生过程和疾病特征有了更深入的了解,sAML 和 tAML 的治疗仍然是一个未被满足的医疗需求,尤其是对于 TP53 突变AML。
参考文献
Semin Hematol . 2025 Jun 30:S0037-1963(25)00032-0. doi: 10.1053/j.seminhematol.2025.06.005.

