青少年起病的成人型糖尿病:基因检测助力精准诊断、疾病管理及遗传咨询
时间:2025-09-03 12:11:18 热度:37.1℃ 作者:网络
青少年起病的成人型糖尿病(MODY)属于单基因糖尿病(MD)这一异质性疾病群体,其特征为胰腺β细胞非免疫性功能障碍。目前,MODY的诊断对临床医生而言仍是一项挑战:许多病例被误诊为1型或2型糖尿病(T1DM/T2DM),且超过80%的病例尚未得到确诊。随着现代技术的应用,在阐明包括MODY在内的单基因糖尿病的分子机制与异质性病因方面已取得重要进展。本研究旨在为一组临床疑似单基因糖尿病的早发糖尿病/糖尿病前期患者,筛选与MODY相关的基因变异。基于下一代测序(NGS)技术的基因检测,要么使用单基因糖尿病的基因panels,要么通过分析整个外显子组(全外显子组测序)进行。GKC-MODY 2是最常见的变异类型,但罕见的KCNJ11-MODY 13,特别是HNF4A-MODY 1也被识别出来。本研究强调了基因检测在早期诊断、MODY亚型区分及遗传咨询中的重要性,并阐述了基因型-表型的相关性(尤其与疾病临床进展和个体化治疗相关),同时也突出了家庭背景下每位患者的个体特征。
研究背景
青少年发病的成人型糖尿病(MODY)属于单基因糖尿病(MD)这一异质性疾病群体,其核心特征为胰腺β细胞功能障碍。单基因糖尿病还包括新生儿糖尿病(ND)、母系遗传糖尿病伴耳聋,以及糖尿病相关综合征(如沃尔夫拉姆综合征(Wolfram syndrome)、巴德特-比德尔综合征(Bardet–Biedl syndrome)、沃尔科特-赖利森综合征(Wolcott–Rallison syndrome)和米切尔-莱利综合征(Mitchell–Riley syndrome))。
根据2008年提出的诊断标准,MODY的定义需满足以下条件:早发非胰岛素依赖性糖尿病(通常发病年龄<25岁);常染色体显性遗传(家族中至少有1名其他患病成员,多表现为跨代遗传);胰腺β细胞非免疫性功能障碍(无自身抗体),且伴随原发性胰岛素分泌缺陷;无代谢综合征。
在儿童、青少年及青年人群中,MODY的患病率呈持续上升趋势,约占所有糖尿病(DM)病例的1%~6%。目前,MODY的诊断对临床医生而言仍是一大挑战:许多病例被误诊为1型或2型糖尿病,且超过80%的病例尚未得到确诊。
MODY与常见类型糖尿病在表型上存在重叠,这可能导致诊断错误。例如,MODY患者可能像1型糖尿病患者一样为体重偏轻的年轻人,但无需胰岛素治疗且无胰腺β细胞自身抗体;也可能表现出类似2型糖尿病的特征,如合并肥胖。此外,1型糖尿病、2型糖尿病与MODY患者均可能存在糖尿病家族史;部分MODY患者甚至无糖尿病家族史,其原因可能是新发突变,或患者父母及其他亲属的糖尿病病史信息不准确。对MODY的正确诊断对于制定治疗方案至关重要,因为适当的治疗方式取决于疾病的病因(例如,对于HNF1A和HNF4A型MODY,采用口服磺酰脲类药物治疗与对1型糖尿病患者使用胰岛素治疗相比有所不同),但这一诊断只能通过分子基因学检测来确认。因此,误诊可能导致治疗不当。在人群整体肥胖率上升的背景下,MODY与2型糖尿病的临床鉴别诊断难度增加,这也进一步凸显了基因检测对实现正确诊断的重要性。此外,基因检测资源获取的不平等,可能是导致MODY患者诊断延迟或误诊的一个重要因素。
1990年后,NGS等现代技术逐渐应用——包括靶向基因panel检测、全外显子组测序(WES)和全基因组测序(WGS)——为阐明包括MODY在内的单基因糖尿病的复杂分子机制与异质性病因奠定了重要基础(图1)。
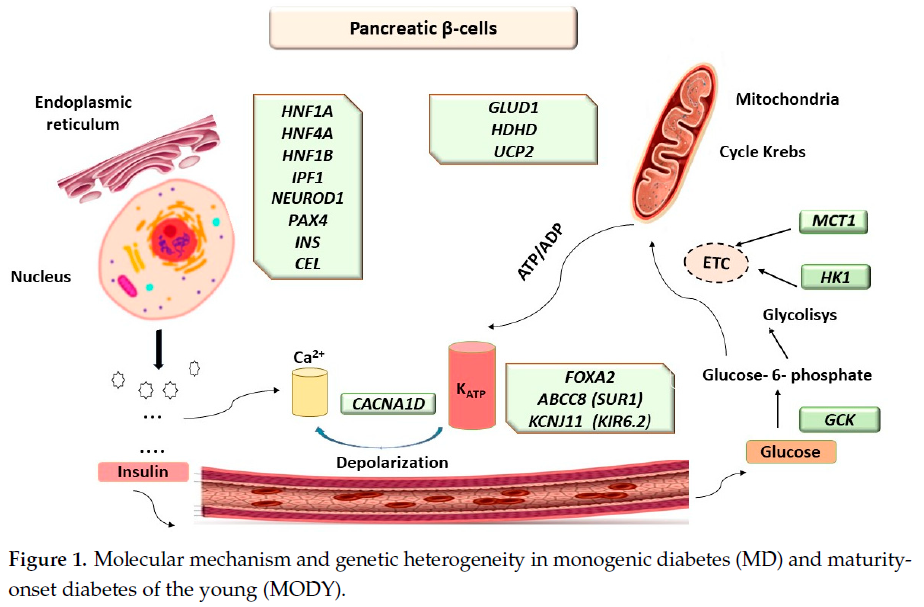
图1
至此,研究已发现14个与MODY相关的基因及位点,这使得根据基因病因对MODY进行重新分类成为可能(所有类型的突变均呈常染色体显性遗传)(表1)。
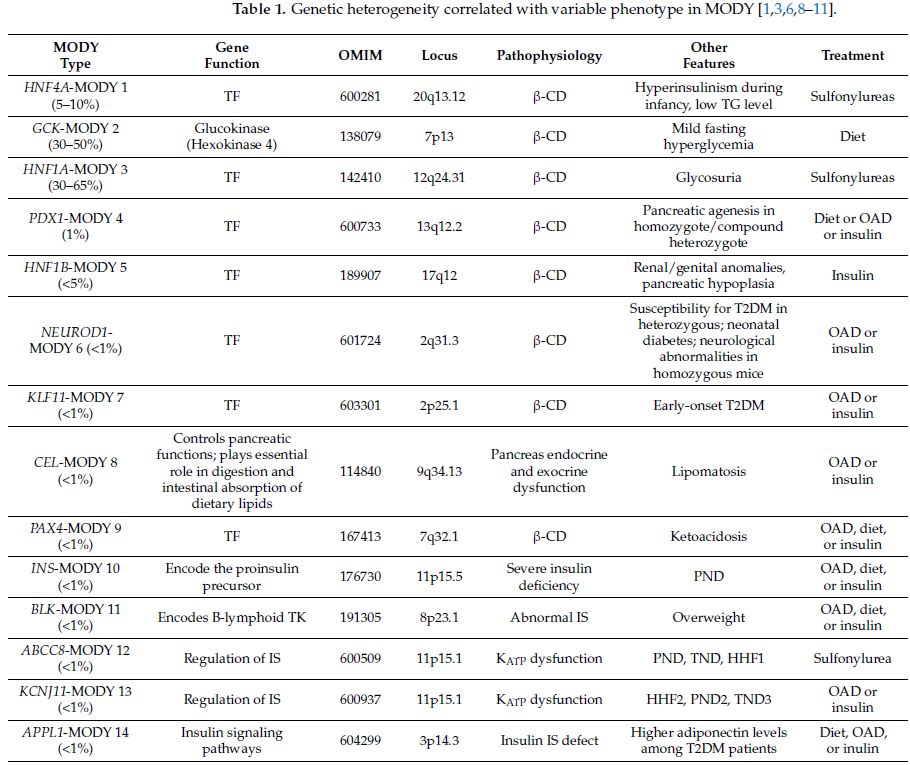
表1
在欧洲,MODY是最常见的单基因糖尿病类型,约占所有糖尿病病例的2%~5%。在高加索人群中,HNF1A(MODY 3型)、GCK(MODY 2型)、HNF4A(MODY 1型)和HNF1B(MODY 5型)基因的突变导致了超过95%的MODY病例。研究显示,欧洲人群与亚洲人群中检测到的基因变异谱存在差异;此外,欧洲不同人群中MODY的患病率也存在差异,且这种差异与基因病因相关。其中,GCK-MODY(2型)和HNF1A-MODY(3型)约占所有MODY病例的30%~60%,在意大利、法国、德国、西班牙和美国,GCK-MODY的患病率相对更高。在英国,最常见的MODY类型是HNF1A-MODY 3型(占比21%~64%);而在法国,GCK-MODY 2型的检出率为8%~63%。在MODY病例中,HNF4A(1型)和HNF1B(5型)变异的检出率约为10%;其他类型的MODY较为罕见,仅在少数家族中被报道。
ABCC8基因编码磺脲类受体1(SUR1),KCNJ11基因编码内向整流钾通道(Kir6.2),这两种蛋白均为胰腺β细胞中ATP敏感性钾通道(KATP)的亚基(图1、表1)。ABCC8和KCNJ11基因的致病性变异是永久性和暂时性新生儿糖尿病(TND/PND)的常见病因。少数情况下,这两个基因的突变也会导致儿童或青年起病的糖尿病,分别归类为ABCC8-MODY 12和KCNJ11-MODY 13。
目前认为,仍有大量MODY病例未被确诊,原因是在已知的14个MODY相关基因中未检测到突变,这类病例被归为MODY-X亚型。研究推测,该亚型可能涉及尚未被发现的额外基因或位点。对于这类病例,WES或WGS等扩展检测手段可能提供更多信息,从而阐明其病因。值得注意的是,部分导致MODY的基因突变还可能与其他类型的糖尿病相关(即存在等位基因异质性和位点异质性,如NEUROD1、KLF11、INS、ABCC8、KCNJ11和APPL1基因)(表1)。
对患者进行分子基因学检测具有重要意义:通过检测可明确突变类型,进而确认MODY诊断、区分亚型、预测可能的临床病程,并为患者的治疗方案提供指导。
HNF4A-MODY 1型和HNF1A-MODY 3型的一线治疗药物为磺脲类药物。大多数GCK-MODY 2型患者无明显症状,血管并发症风险较低,因此通常无需药物治疗。
遗传咨询是MODY患者管理中的关键环节。由于MODY呈常染色体显性遗传,先证者一级亲属的疾病再发风险为50%。对患病个体的家庭成员进行基因检测,可实现突变携带者的症状前诊断,进而为其采取预防措施(包括定期血糖监测、早期诊断和适宜治疗)提供依据。
本研究旨在为一组临床疑似单基因糖尿病的早发糖尿病/糖尿病前期患者,筛选与MODY相关的基因变异;同时阐述基因型-表型的相关性,重点分析家庭背景下患者个体特征与所检测到的突变类型的关联(尤其涉及疾病临床进展和个体化治疗方面)。
研究结果
对8例MODY患者进行扩展性基因检测(单基因糖尿病基因panel检测和WES)后发现,其中6例患者携带GCK-MODY 2型的突变,另外2例患者携带KCNJ11-MODY 13型和HNF4A-MODY 1型的罕见突变(表2)。在所有8例MODY患者中检测到的突变均为致病性突变,其中6个为错义突变,1个为无义突变,另有1个为杂合性剪接供体突变(表1)。
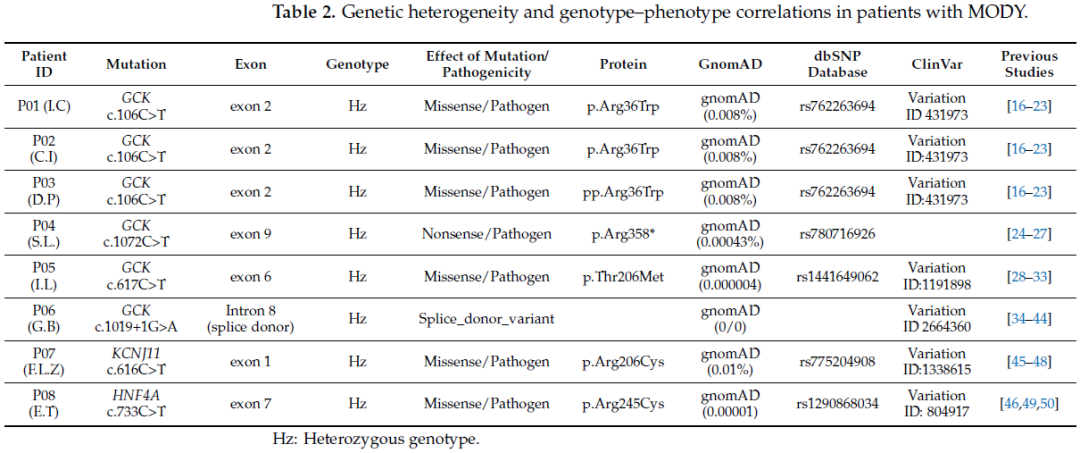
表2
在3例GCK-MODY 2型患者中,均检测到位于GCK基因第2外显子的杂合性错义突变GCK c.106C>T(对应蛋白改变为p.Arg36Trp)(图2)。1例MODY患者检测到位于第6外显子的另一种杂合性错义突变GCK c.617C>T(p.Thr206Met),另有1例患者检测到位于第9外显子的无义突变GCK c.1072C>T(p.Arg358*)。此外,在1例患者中检测到杂合性剪接供体突变GCK c.1019+1G>A(表3、图2)。位于KCNJ11基因第1外显子的错义突变KCNJ11 c.616C>T(p.Arg206Cys)被认为是MODY的罕见突变;在多数情况下,KCNJ11基因突变与永久性及暂时性新生儿糖尿病(TND/PND)相关,极少在儿童或青年糖尿病患者中检测到。在另1例患者中检测到另一种罕见错义突变,该突变位于HNF4A基因第7外显子,具体为HNF4A c.733C>T(p.Arg245Cys)。其他HNF4A基因变异与某些类型的迟发性非胰岛素依赖型糖尿病(迟发性NIDDM)有关(OMIM编号:125853)。
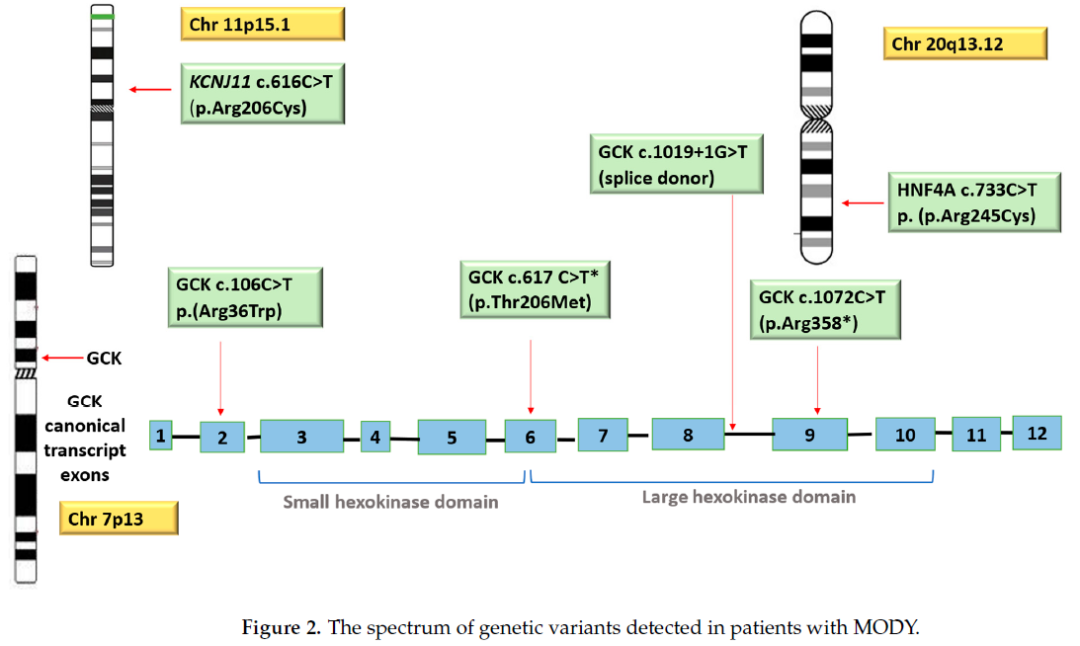
图2
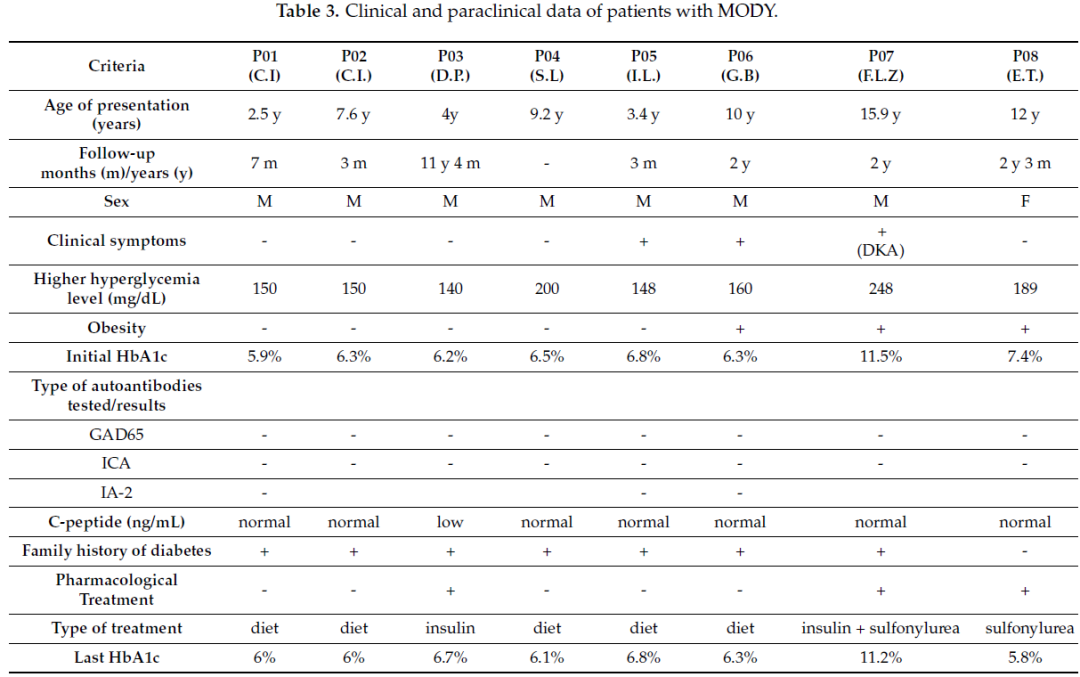
表3
MODY患者的临床病史资料及生化指标数据如表3所示。患者确诊时的年龄范围为2.5~15.9岁;8例患者中7例为男性,仅1例为女性(病例P08,姓名缩写E.T.)。8例患者中有7例存在DM家族史阳性;病例P08的DM家族史为阴性,其携带的突变极可能为新发突变(图3)。
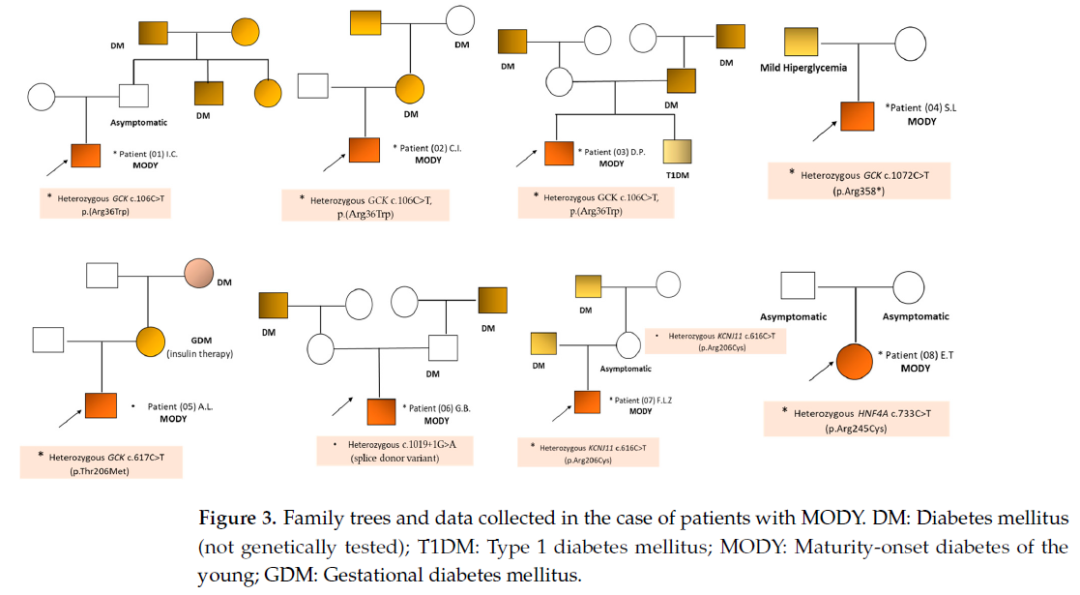
图3
为排除免疫介导的1型糖尿病(T1D),所有糖尿病前期/糖尿病患者均在初始阶段检测了血浆C肽水平及至少两种抗体:抗谷氨酸脱羧酶65抗体(抗-GAD65),以及抗酪氨酸磷酸酶相关胰岛抗原2抗体(抗-IA2)或抗胰岛细胞抗体(抗-ICA)中的任意一种(表3)。
对于存在糖尿病家族史阳性、抗体检测阴性且糖化血红蛋白(HbA1c)水平即使仅轻度升高的患者,均需进行基因检测。
生化指标(高血糖、血清C肽水平、HbA1c、抗-GAD65抗体、抗-ICA抗体及抗-IA2抗体)的变化情况如表3所示。患者的血糖最高值范围为140~248 mg/dL(毫克/分升),HbA1c水平范围为5.9%~11.5%(表3)。8例患者中有2例出现临床症状(多尿、多饮及代谢综合征),其中1例还合并酮症酸中毒(DKA)。3例患者(P06、P07、P08)存在肥胖。5例患者(P01、P02、P04、P05、P06)无需药物治疗;仅1例患者(P03)在基因检测后仍继续使用此前已开始的胰岛素治疗;1例患者(P07)接受胰岛素联合磺脲类药物治疗并获益;1例患者(P08)接受磺脲类药物联合饮食控制治疗(表3)。
讨 论
在本研究纳入的MODY患者中,绝大多数(6/8)通过分子基因检测(MODY基因panel检测或WES)发现存在GCK突变(即MODY 2型)。其中3例患者(P01、P02、P03)检测到相同的致病性突变GCK c.106C>T(表2、图3)。6例GCK-MODY 2型患者中,有5例(P01、P02、P04、P05、P06)无需药物治疗,仅建议其进行饮食控制与血糖监测;另有1例患者(P03),初始同样建议饮食控制,但由于其HbA1c水平从6.2%升至6.7%,故在基因检测后仍维持此前已启动的基础胰岛素治疗。
第1例患者(P01,I.C.,2岁)在常规检查中,被发现血浆葡萄糖水平升高(150 mg/dL),且HbA1c水平介于5.9%~6.1%之间,无其他临床症状(表3)。抗-GAD65与抗-IA2均为阴性。该患儿的特殊情况为存在精神运动性兴奋发作。家族史方面,患儿父亲的兄弟姐妹及祖父母均确诊糖尿病(未接受基因检测)(图3)。
第2例患者(P02,C.I.,7岁8个月)因空腹血浆葡萄糖水平升高(最高值130 mg/dL)接受评估,同时伴HbA1c为6.3%、餐后2小时血糖为150 mg/dL,且无自身免疫相关指标(未检测到胰岛自身抗体)。家族史呈阳性:患儿母亲曾患妊娠期糖尿病(GDM),需接受胰岛素治疗;患儿外祖父确诊糖尿病(考虑为2型糖尿病),需接受口服降糖药(OA)治疗(均未接受基因检测)(图3)。该患儿还患有注意缺陷多动障碍(ADHD),目前接受托莫西汀(商品名:择思达)治疗。通过Blueprint Genetics实验室的WES检测,发现其携带杂合致病性突变GCK c.106C>T(p.Arg36Trp)(表2)。
第3例患者(P03,D.P.,目前15岁6个月)于2013年(时年4岁)被诊断为糖尿病前期,依据为血浆葡萄糖平均水平150 mg/dL、HbA1c为6.7%,且血浆C肽水平为0.51 ng/dL(表3)。初始建议其进行饮食控制,后期启动基础胰岛素治疗。家族史呈阳性:患者父亲及祖父母均确诊糖尿病(考虑为2型糖尿病),需接受口服降糖药治疗(均未接受基因检测)。此后,患者兄长被诊断为1型糖尿病(T1DM),且胰岛素需求量增加(图3)。在尝试逐步停用胰岛素治疗的过程中,观察到患者血糖及HbA1c水平轻度升高,应其父母要求,最终决定维持最小剂量的基础胰岛素治疗。此外,该患儿还存在骶尾部脊柱裂,但无需手术治疗。对患者及其兄长进行的基因检测(Invitae单基因糖尿病panel检测)显示,患者携带GCK c.106C>T突变,而其兄长未检测到该突变。
杂合突变GCK c.106C>T(p.Arg36Trp)在gnomAD数据库中记录于4例杂合子患者,同时也存在于ClinVar数据库(编号431973)。所有使用的计算机模拟工具均预测该突变为致病性突变。GCK c.106C>T突变最早在一个法国家庭中被报道与常染色体显性高血糖相关,且该家庭中的突变为新发突变(PMID: 8168652 )。此后,该突变在多例MODY患者或轻度高血糖患者及家庭中被报道(PMID: 25555642、35592779、28012402、33477506)。
Fendler等人在68例波兰GCK-MODY患者中,有11例检测到该突变(PMID: 21521320)。Valentínová等人在一个斯洛伐克MODY家庭中报道了该突变,该家庭中所有患病成员均为杂合基因型(PMID: 22493702)。一项功能研究显示,p.Arg36Trp突变蛋白与野生型蛋白具有相似的动力学特性,且在热稳定性方面与野生型蛋白无差异(PMID: 10426385)。
第4例患者(P04,S.L.,9岁3个月)因检测到血浆葡萄糖水平为200 mg/dL(无其他临床症状)接受评估。临床检查未发现任何病理性指标,且无胰岛素抵抗相关体征(肥胖、黑棘皮症或代谢综合征)。抗-GAD65与抗-IA2抗体均为阴性,血浆C肽水平正常。通过Blueprint Genetics(BpG)MODY基因panel进行NGS检测,发现其携带致病性突变GCK c.1072C>T(p.Arg358*),该突变为无义突变,可导致GCK蛋白第358位的精氨酸被提前终止密码子替代,进而影响蛋白的一个功能结构域。该序列变异在GCK基因中产生了提前翻译终止信号(p.Arg358*),预计会导致蛋白产物缺失或功能异常。已知GCK基因中的功能缺失性变异(LOF)具有致病性(PMID: 10754480、14578306、24323243、25015100)。
该突变在人类基因突变数据库(HGMD)中被描述为与MODY 2型相关的致病性变异(编号CM002026),同时存在于dbSNP数据库(编号rs780716926)及人群频率数据库gnomAD(频率0.00043%)中。生物信息学预测工具MutationTaster评估认为该变异具有致病性。在查阅的科学文献中,该患者携带的这一变异在西班牙MODY患者队列的2个病例中被报道(PMID: 10754480)(ESPE摘要(2019)92 RFC1.4)。家族史显示,患者父亲偶发中度高血糖,未接受任何治疗。
第5例患者(P05,A.L.,3岁5个月)起病时出现糖尿病特异性症状(多尿、多饮、体重下降),同时伴血浆葡萄糖水平升高(平均148 mg/dL)、HbA1c为6.8%。血浆C肽水平正常,抗-GAD65与抗-IA2抗体均为阴性,排除1型糖尿病(T1DM)可能。家族史呈阳性:患者母亲曾患需胰岛素治疗的妊娠期糖尿病,外祖母确诊2型糖尿病(T2DM)。通过Invitae单基因糖尿病panel检测,发现其GCK基因存在杂合致病性变异c.617C>T,该变异导致GCK蛋白第206位密码子的苏氨酸被蛋氨酸替代(p.Thr206Met)。苏氨酸残基具有高度保守性,且苏氨酸与蛋氨酸之间存在中度理化性质差异。在进行基因检测时,该基因变异被认为是新发变异,未在文献中报道;此后,该变异在国际数据库中被报道,并被归类为致病性变异(ClinVar,变异编号1191898;gnomAD;dbSNP数据库,编号rs1441649062)。多项研究在常染色体显性遗传的MODY患者中检测到该变异(PMID: 34440516、31216263、28726111、24606082、19790256、16173921)。
用于预测错义变异对蛋白质结构与功能影响的算法(SIFT、PolyPhen-2、Align-GVGD)显示,该变异可能破坏编码蛋白的功能。实验研究表明,该变异会影响GCK蛋白的功能(PMID: 16173921),这些结果支持将该变异归类为致病性变异。该患者的特殊情况为:存在下腔静脉发育不全(心下段缺失),且遗传性易栓症相关检测呈阳性——携带亚甲基四氢叶酸还原酶(MTHFR)A1298C、纤溶酶原激活物抑制剂-1(PAI-1)675 4G/5G及内皮细胞蛋白C受体(EPCR)A2/A3杂合变异,这些变异与深静脉血栓形成易感性相关。
第6例患者(P06,G.B.,目前10岁)于8岁时起病,当时检测到高血糖(160 mg/dL),伴HbA1c为6.3%、C肽水平正常,且抗-GAD65与抗胰岛细胞抗体(抗-ICA)均为阴性。该患儿同时存在肥胖与继发性血脂异常。家族史呈阳性:外祖父母与祖父母均确诊糖尿病(未接受基因检测)(图3)。通过Blueprint Genetics(BpG)MODY基因panel进行NGS检测,发现其携带GCK基因杂合剪接供体变异c.1019+1G>A(表3)。
该变异在大型参考人群数据库gnomAD中未被检出(该数据库包含超过12万个外显子组与1.5万个基因组数据,旨在排除患有严重儿科疾病的个体),但在ClinVar数据库中被报道(变异编号2664360)。该变异替换了经典剪接位点内的一个核苷酸,因此可能导致异常剪接。若不进行转录研究,无法验证该变异对编码蛋白的影响;但预测该变异会导致外显子框内跳跃,进而使蛋白缺失52个氨基酸。已有研究报道该外显子区域存在致病性错义变异(PMID: 34826540、36257325、14517956、15928245),表明该蛋白区域具有重要功能。在多例GCK相关表型患者中,均报道过该GCK c.1019+1G>A变异(PMID: 15928245、28012402、32533152、31638168、31529753)。
此外,已有研究在GCK相关疾病患者中报道了其他影响同一剪接供体位点的变异,包括c.1019+5G>C、c.1019+5G>A、c.1019+3dupG、c.1019+2T>G、c.1019+2T>C、c.1019+2T>A及c.1019+1delG(PMID: 14578306、19564454、31638168、29056535、31638168、27908292)。
第7例患者(P07,F.L.Z.)于15岁9个月时确诊糖尿病,当时出现多尿(伴夜尿)、多饮症状,且血糖水平升高。此外,该患者存在重度肥胖,体重指数(BMI)为37.4 kg/m²(较对应身高的标准体重超出45~50 kg)。糖尿病诊断通过生物学检查确认(检测到HbA1c为11.5%),并启动基础-餐时胰岛素治疗(BBT),使用超短效胰岛素类似物与甘精胰岛素300 U。
患者血浆C肽水平为2.62 ng/dL(正常),抗-GAD65与抗-ICA抗体均为阴性(表3)。考虑到患者家族史呈阳性(其父近期确诊2型糖尿病),且无自身免疫相关体征,故怀疑其为单基因糖尿病(MD)。对患者家庭成员进行分子检测(Invitae单基因糖尿病panel检测)发现,患者及其母亲(无症状)均携带KCNJ11基因杂合致病性变异c.616C>T(p.Arg206Cys),而患者父亲与妹妹未检测到该突变。家族史显示,患者外祖父也曾确诊糖尿病,且需接受胰岛素治疗。
基于基因检测结果,重新评估了治疗方案,将胰岛素治疗替换为最小剂量的磺脲类药物(格列齐特)。患者接受口服降糖药(OA)治疗2个月后复诊,结果显示:肥胖加重(体重137 kg,BMI 38 kg/m²),伴临界性高血压,血糖控制不佳(HbA1c 11.2%)。随后增加磺脲类药物剂量,并联合基础胰岛素治疗(国际非专利名称:德谷胰岛素)与低热量饮食。
位于KCNJ11基因第1外显子的c.616C>T变异为错义突变,可导致KCNJ11蛋白第206位密码子中,具有碱性与极性的精氨酸被中性且弱极性的半胱氨酸替代(p.Arg206Cys)。该变异存在于国际数据库中(rs775204908,gnomAD数据库中频率0.01%;ClinVar编号1338615),且在多项研究中被报道存在于常染色体显性遗传的家族性高胰岛素血症患者中(PMID: 25555642、30026763、12524280、31464105、23348805)。
用于预测错义变异对蛋白质结构与功能影响的算法(SIFT、PolyPhen-2、Align-GVGD)显示,该变异可能破坏蛋白功能;实验研究证实,该突变会破坏胰腺β细胞中ATP敏感性钾通道(KATP通道)Kir6.2亚基蛋白的第206位精氨酸残基(p.Arg206)(PMID: 12524280)。
已有研究确定,其他破坏该残基的变异具有致病性(PMID: 31464105;Invitae),这表明该残基具有临床意义,破坏该残基的变异可能引发疾病。基于上述原因,该变异被归类为致病性变异。
考虑到患者母亲携带相同突变但无糖尿病特异性症状(无症状),因此对该突变在其体内表现出的可变表达性与不完全外显性进行了探讨。
最后1例患者(P08,E.T.,女性,12岁)因检测到血浆葡萄糖水平升高(最高达189 mg/dL)且伴糖尿而接受评估。临床检查显示,该患者同样存在肥胖(BMI 24.6 kg/m²,位于第97百分位)。生物学检查显示,患者血浆HbA1c水平为7.4%,血浆C肽水平正常,且未检测到抗-GAD65与抗-ICA抗体。尽管患者糖尿病家族史呈阴性,但仍建议其进行基因检测(Invitae单基因糖尿病panel检测)。检测结果显示,患者HNF4A基因第7外显子存在杂合致病性变异c.733C>T(p.Arg245Cys),该变异在ClinVar数据库中可查(变异编号804917)。该错义突变导致HNF4A蛋白第245位密码子中,具有碱性与极性的精氨酸被中性且弱极性的半胱氨酸替代(p.Arg245Cys)。
由于gnomAD数据库中关于该变异的数据不足,无法估算其在普通人群中的频率。HNF4A c.733C>T变异在家族性高胰岛素血症和/或MODY患者中均有报道(PMID: 30026763、23348805、31957151;Invitae)。至少有1例患者的该突变为新发突变,其家族史呈阴性。
这一情况表明,即使在无糖尿病阳性家族史的患者中,若其未出现1型糖尿病(T1DM)相关自身免疫标志物,仍可怀疑MODY诊断。
用于预测错义变异对蛋白质结构与功能影响的算法(SIFT、PolyPhen-2、Align-GVGD)显示,该变异可能具有致病性。
考虑到患者父母均无症状且基因检测结果为阴性,该患者极可能携带新发突变,但无法排除父母一方存在胚系嵌合现象的可能性。
患者初始治疗方案为饮食控制联合磺脲类药物(格列齐特)。由于在口服降糖药(OA)最小剂量下,患者仍出现低血糖发作,故决定停用药物治疗。仅通过饮食控制,患者血浆糖化血红蛋白(HbA1c)水平维持在5.8%。未能对所有先证者的家庭成员(尤其是已确诊糖尿病的成员)均进行基因检测。仅在2个家庭(P07、P08)中,实现了对所有一级亲属的基因检测;而在患者P03的家庭中,仅对其兄长进行了检测,未检测父母。研究者认为,基因检测具有必要性,尤其对于GCK-MODY 2型患者家庭——通过检测可重新评估治疗方案,甚至停用降糖药物。
研究者认为,本研究的局限性在于分析病例数量较少,研究结果无法推广至摩尔多瓦地区所有疑似MODY的患者。造成这一局限的原因包括:无法为所有符合MODY临床标准的患者及其家庭成员提供分子检测(基因检测费用由家庭承担);此外,在部分病例中,难以获取关于糖尿病家族史的最准确信息。
然而,本研究结果与其他欧洲研究(纳入大量MODY患者队列)的结果一致。已知MODY的患病率在不同人群中存在差异,且不同人群中的致病基因变异可能因种族而异。
例如,Passanisi等人在565例儿童及青少年糖尿病患者中,发现37例(6.5%)为MODY(其中68例患者接受了单基因糖尿病基因检测)。在这些MODY患者中,30例(81%)为GCK-MODY 2型,5例(13.5%)为HNF1A-MODY 3型,2例(5.4%)为HNF4A-MODY 1型。患者年龄范围为0~16岁,平均年龄9.1岁。
Avelos等人在46个接受分析的葡萄牙家庭中,发现23例(50%)患者为MODY。其中12例患者携带GCK基因变异,8例携带HNF1A基因突变,3例携带HNF4A基因变异。在这些变异中,16个为错义突变,2个为无义突变,1个为移码突变,1个为同义变异。
在一项针对卡塔尔人群的研究中,纳入了37例符合MODY临床标准的患者,在24例患者中检测到10个错义突变。检测到的变异大多位于HNF1A基因,其中最常见的是rs587778397(p.Arg177Trp)。5例患者携带BLK基因变异(rs766934515),4例患者携带GCK基因突变。此外,在HNF4A、GCK、KLF11及ABCC8基因中还检测到其他罕见突变。
Ben Kelifa等人对23例无亲缘关系、符合MODY临床标准的突尼斯患者进行了分析。通过测序及MLPA,对患者GCK、HNF1A、HNF4A及INS的突变情况进行了检测。在3例先证者(13.05%)中检测到GCK及HNF4A基因突变,未在任何患者中检测到HNF1A及INS基因突变。仅在1个家庭(先证者及其母亲)中检测到已知的HNF4A c.-169C>T变异;在2例无亲缘关系的患者中,发现了新的HNF4A c.-457C>T突变。由于这两个家庭的成员拒绝基因检测,无法验证该突变在家庭中的遗传分离情况。在另外2例无亲缘关系的患者中,发现了新的GCK c.457C>T突变。作者得出结论:尽管采用了严格的临床标准筛选患者,但已知与MODY相关的常见基因的突变,并不能解释大多数突尼斯患者的发病原因。这提示在突尼斯人群中,可能存在其他尚未被发现的致病基因,参与MODY的发病机制。
Bario等人在22例西班牙裔儿童患者中,发现了与MODY相关的3个基因中的14个突变。其中9个为新突变,且所有突变均在家族系谱中与MODY临床表型共分离。GCK-MODY 2型突变最为常见(占病例的41%):其中7个为新发突变(R369P、S411F、M298K、C252Y、Y108C、A188E、S383L),2个为已知突变。在4例患者(18%)中检测到HNF1A-MODY 3型变异(包括新突变R27G)。在1个家庭(4%)中检测到HNF4A IVS5-2delA(MODY 1型)变异,该变异在西班牙人群中为首次报道。
MODY面临的新挑战及未来发展方向如下:
从临床角度而言,提高诊断方法的准确性有助于区分MODY与其他类型糖尿病(T1DM/T2DM),从而避免不必要的胰岛素或磺脲类药物治疗——这类治疗可能对患者健康产生不利影响。未来,需针对单基因及多基因糖尿病开展转化生物学与整合基因组学相关的新研究,这将为揭示糖尿病病理生理过程中的复杂分子机制及糖尿病治疗提供新视角。
NGS技术在MODY诊断中的应用,是明确MODY致病因素的关键——该技术可识别已知致病基因或新候选基因,而这些基因正是胰腺β细胞功能障碍的根本原因。因此,NGS技术或可阐明部分家族性或非典型早发型糖尿病的发病原因。
转录组学分析可提供与MODY发病相关分子机制的重要信息。代谢组学分析或可成为研究并区分MODY与其他类型糖尿病的另一方向;此外,多能干细胞(PSCs)的应用或有助于揭示包括MODY在内的不同类型糖尿病的潜在分子机制。
MODY患者的遗传咨询:
遗传咨询在MODY患者管理中发挥重要作用。在本研究分析的队列中,8例患者中有7例存在糖尿病阳性家族史。基于该病的常染色体显性遗传模式,这些患者的一级亲属患病复发风险为50%。患者E.T.(P08)无糖尿病家族史,由于在其健康父母中未检测到该儿童携带的基因变异,故该患者极可能携带HNF4A基因新发突变,但无法排除父母一方存在胚系嵌合现象的可能性。针对MODY患者及其家庭成员的遗传咨询必须遵循生物伦理原则。大多数MODY患者为儿童或青少年,因此关注患者及其家庭的长期健康至关重要。需以清晰、恰当且非指令性的方式,向患者及家属告知疾病相关信息,包括疾病诊断(含基因检测确认结果)、疾病进展,以及MODY患者其他家庭成员或后代的发病风险。当医生建议对儿童进行基因检测,或需向其他可能存在MODY高风险的家庭成员披露儿童的疾病相关信息(包括基因检测结果)时,儿童父母可能会感到不安。
对患者家庭成员进行基因检测,可实现突变携带者的症状前诊断——这类携带者需采取预防措施,包括定期血糖监测、早期诊断及规范治疗。
结 论
综上所述,本研究对一组临床疑似MD/MODY的患者,采用特定基因panel的靶向基因测序技术,部分病例辅以WES,旨在明确其涉及的基因变异(包括已知变异与新变异)。
GCK-MODY 2型是本研究中检出频率最高的变异类型,但同时也发现了罕见的KCNJ11-MODY 13和HNF4A-MODY 1型。本研究强调,在单基因糖尿病诊断中应用高性能分子技术(NGS)及基因panel检测或WES,是准确识别患者及其他家庭成员MODY亚型的关键因素。若不进行基因检测,考虑到本研究纳入的多数患者发病年龄较小、部分患者无相关临床症状,且部分病例无阳性家族史,疾病诊断可能会延迟。精准医疗的应用(包括将基因检测纳入MODY等单基因糖尿病的诊断流程),应成为此类疾病患者管理中的标准方法——尤其是因为基因变异、患者表型与所用治疗方案之间存在相关性。
明确与种族和民族相关的单基因糖尿病基因生物标志物,将是未来研究面临的挑战。
将基因检测推广至更大量、更多样化的人群,对基因变异开展功能研究,同时为MODY基因检测建立完善的伦理框架与咨询规范,或有助于实现基于基因病因的早期诊断与个体化治疗。为MODY患者制定诊疗指南,可推动基因检测的普及。此外,未来研究应探索遗传咨询的最佳实践模式,尤其是针对家庭成员存在MODY高发病风险的情况。通过基因检测,可对携带MODY相关基因变异的个体进行症状前诊断,这类个体需采取预防措施,包括血糖监测与饮食控制。
“遗传病全外显子组检测”项目,针对单基因遗传病,全外显子检测2万+个目标基因(包含线粒体),覆盖点突变(SNV)、小片段插入缺失(Indel)、以及预测基因扩增(CNV)等变异类型,评估单基因遗传病及遗传性肿瘤的发病风险。
参考文献:
Butnariu, L.I.; Bizim, D.A.; Oltean, C.; Rusu, C.; Pânzaru, M.C.; Păduraru, G.; Gimiga, N.; Ghiga, G.; Moisă, Ș.M.; Țarcă, E.; et al. The Importance of Molecular Genetic Testing for Precision Diagnostics, Management, and Genetic Counseling in MODY Patients. Int. J. Mol. Sci. 2024, 25, 6318. https://doi.org/10.3390/ijms25126318

