华中大李一伟、刘笔锋教授Cell Systems: 开发肿瘤-脂肪组装体新技术,揭示脂肪肿瘤力学相互作用与癌症侵袭机制
时间:2025-08-27 12:09:35 热度:37.1℃ 作者:网络
北京时间2025年8月8日,华中科技大学生命科学与技术学院李一伟教授和刘笔锋教授团队在Cell Systems《细胞·系统》期刊发表了题为“Tumor-Adipose Assembloids Reveal Cell-Fate-Transition-Triggered Multistage Collective Invasions”的研究文章。该论文可以通过此链接https://authors.elsevier.com/a/1lZlf8YyDfuZU7。

论文通讯作者是李一伟教授和刘笔锋教授,论文共同第一作者是博士生雷梦成和李家硕。该论文开发了一种创新的肿瘤-脂肪组装体系统,为癌症侵袭机制研究提供了前所未有的新视角。这项突破性工作成功架起了生物过程与物理行为在肿瘤进展中相互作用的桥梁,代表了癌症研究方法学和治疗靶点发现的重大进展。
癌症侵袭是肿瘤进展过程中最复杂、最致命的环节之一,涉及癌细胞与其微环境之间错综复杂的相互作用。虽然科学界早已认识到物理力学和生物细胞命运转换在肿瘤侵袭中均发挥着关键作用,但二者在时间维度上的耦合机制一直是个谜。
肥胖与癌症的关联已成为全球性健康危机,脂肪组织作为肿瘤微环境的重要组成部分发挥着关键作用。此前研究表明,肿瘤生长产生的机械压迫可导致脂肪细胞去分化,并可能促进癌症进展。然而,脂肪细胞促进肿瘤侵袭的确切机制以及这些相互作用的时间轴,由于现有研究模型的局限性而难以深入研究。
传统研究方法面临重大挑战:二维细胞培养系统虽然允许直接观察,但缺乏理解组织水平行为所必需的三维组织结构;而动物模型虽然具有生理相关性,但无法实现对个体细胞行为和命运转换的详细实时追踪。
创新方法:组装体技术突破
为了克服这些局限性,华中科技大学团队开发了肿瘤-脂肪组装体系统,采用了润湿性图案化微芯片技术。这种方法巧妙地解决了一个根本性技术挑战:脂肪细胞在培养基中天然漂浮,而癌细胞下沉,使得以受控方式将它们结合在一起变得困难。
研究人员采用双液滴阵列技术,从相反方向融合每种细胞类型的球体。当两个微芯片面对面对齐时,漂浮的脂肪球体与下沉的肿瘤球体在界面处接触,形成精确控制的肿瘤-脂肪组装体阵列。这种方法学使得在整个培养期间能够实时监测集体形态发生和细胞命运转换。
突破性科学发现
两阶段侵袭机制:研究揭示了肿瘤侵袭通过两个不同阶段发生,每个阶段都受不同生物物理学原理支配:
第一阶段:粘弹性融合(0-48小时)
在初始阶段,团队发现癌细胞和脂肪细胞之间的相互作用可以准确建模为粘弹性液滴之间的接触,类似于高分子材料的行为。使用Johnson-Kendall-Roberts (JKR)理论结合动力学建模,他们表征了融合过程并预测了触发脂肪细胞去分化的内部压应力的产生。
第二阶段:基质驱动侵袭(72小时以上)
在脂肪细胞重编程之后,组装体经历了戏剧性转变。脂肪细胞衍生的肌成纤维细胞出现并开始主动重塑周围的胶原基质,为癌细胞侵袭创造通路。这一阶段的特征是形成延伸到周围组织的侵袭性分支。
细胞命运转换作为侵袭催化剂。最重要的发现之一是确定细胞命运转换作为催化事件,从根本上改变了肿瘤侵袭的物理特性。研究团队观察到脂肪细胞经历两阶段重编程过程:去分化:脂肪细胞失去其特征性脂滴并表达间充质干细胞标志物如CD105;肌成纤维细胞化:从约72小时开始,一些去分化的脂肪细胞进一步转化为表达α-SMA(α-平滑肌肌动蛋白)的肌成纤维细胞。这些肌成纤维细胞被证明是侵袭的关键驱动因子,通过机械力主动招募和重组胶原纤维。
整合素α5:关键干预靶点。研究确定整合素α5作为侵袭过程的关键介质。这种蛋白质特异性地由脂肪细胞衍生的肌成纤维细胞表达,使细胞与细胞外基质之间的机械反馈回路成为可能,从而驱动侵袭。当研究人员使用特异性抗体抑制整合素α5时,成功阻止了侵袭行为并减少了组装体扩张。数学建模显示,阻断整合素α5将侵袭指数从1.05降低到0.65,表明定向驱动力被抑制。这一发现表明整合素α5是在脂肪丰富环境中预防肿瘤侵袭的有希望的治疗靶点。
临床意义和应用
理解肥胖相关癌症风险:这项研究为肥胖增加癌症风险和进展提供了关键的机制性见解。组装体模型证明了肿瘤生长的机械应力如何将脂肪细胞重编程为促癌肌成纤维细胞,创造了加速侵袭的前馈回路。
药物发现平台:组装体系统为药物筛选和治疗开发提供了强大的平台。研究人员现在可以测试针对侵袭过程特定阶段或阻断关键分子相互作用(如整合素α5信号传导)的干预措施。
精准医学应用:该技术可能适用于患者特异性应用,使用患者来源的细胞创建个性化肿瘤模型,用于测试治疗策略和预测治疗反应。
研究团队:
李一伟教授2020年底从麻省理工学院回国加入华中科技大学任教授,专注于细胞物理力学、类器官智能制造和相分离生物技术开发研究。这项工作得到了国家重点研发计划和国家自然科学基金等项目的支持。团队网站:www.yiweililab.com
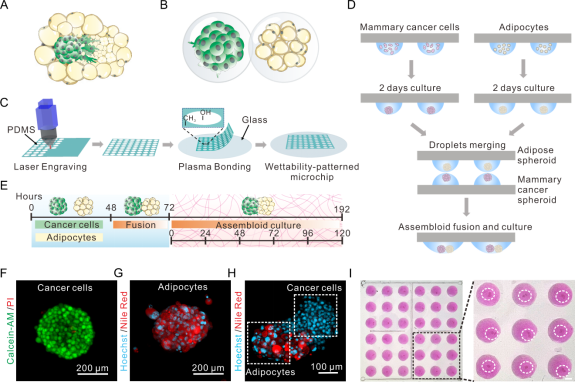
图1展示了肿瘤-脂肪组装体的制备流程、培养及表征。利用激光雕刻技术在PDMS薄膜上雕刻圆形阵列,并通过等离子体键合将其与玻璃基片结合,构建出具有亲疏水图案的阵列芯片。在芯片的亲水区域接种悬浮细胞,可分别形成肿瘤球和脂肪球。随后,采用双液滴阵列对接的方法,将来源不同的细胞球从相反方向融合:当两块微芯片面对面对齐时,漂浮的脂肪球与下沉的肿瘤球在界面处精确接触,从而构建出空间位置可控的肿瘤-脂肪组装体阵列。荧光成像结果表明,该组装体在结构上呈现出鲜明的双区特征:肿瘤区细胞核分布密集,而脂肪区细胞核稀疏,并伴随显著的脂滴信号,二者在保持紧密融合的同时边界清晰可辨。该方法不仅解决了肿瘤细胞与脂肪细胞因物理性质差异而难以稳定组装的技术难点,还使得在整个培养过程中能够实现集体形态发生与细胞命运转换的实时监测。
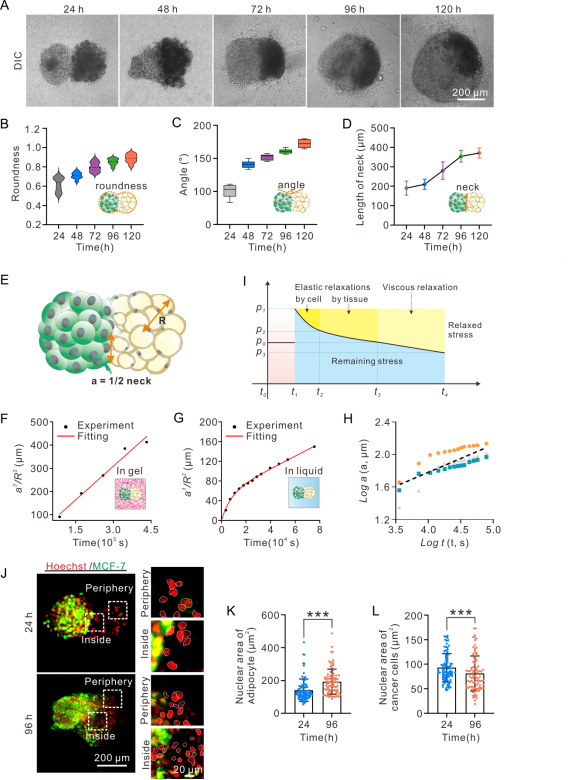
图2展示了肿瘤-脂肪组装体的生物物理特性表征与建模。随着培养时间延长,组装体的圆度增加,融合的颈部半径增大,颈部切线角度增大。使用Johnson-Kendall-Roberts(JKR)理论结合动力学建模,发现在融合过程中,组装体a3/R2值随t的变化与Jelly-Pearl模型预测的达到平衡的动力学密切一致。融合初期(0-48小时)癌细胞和脂肪细胞之间的相互作用可以准确建模为粘弹性液滴之间的接触。结合后,内部压缩应力最初因组装体内部细胞的弹性变形而衰减,随后在组织尺度上发生弹性松弛,并通过粘性松弛进一步消散,最终达到平台状态。对组装体内的脂肪细胞和癌细胞核的形状进行了成像,在组装后前24小时内,非连接区与边缘区细胞的核面积之间无显著差异。组装体培养96小时后,组装体边缘区脂肪细胞的核面积大于连接区。鉴于核结构变化已被提出作为细胞命运转化的标志物,推测细胞转化可能由内部应力引发的核变形触发。
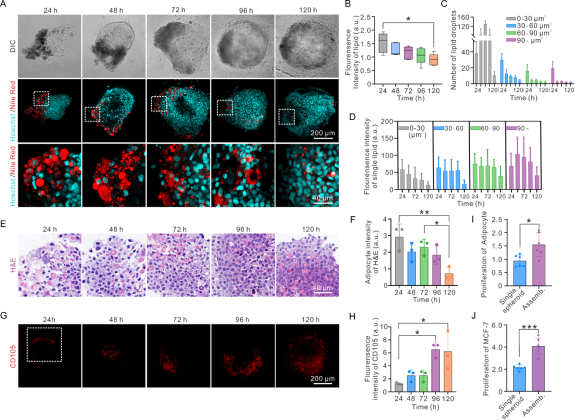
图3描述了组装体中脂肪细胞去分化的过程。随着融合培养时间的延长,组装体中脂滴总荧光强度减弱,脂滴的数量减少,单个脂滴的荧光强度减弱。组装体H&E染色结果显示,脂肪细胞减少。此外,组装体中干细胞标志物CD105表达增强,上述结果共同说明脂肪细胞正在经历转化。此外,与单独培养的脂肪细胞球和脂肪球进行比较,组装体中肿瘤细胞增殖增强,且脂肪细胞恢复部分增殖能力。
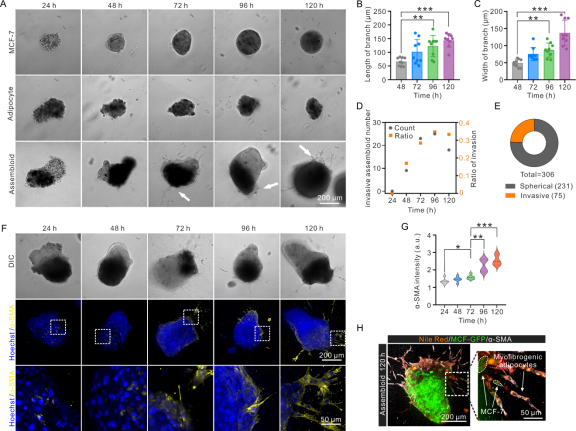
图4描述了组装体中脂肪细胞命运转化及肿瘤细胞的侵袭。与单独培养的脂肪球和肿瘤球相比,只有组装体出现侵袭分支,随着随培养时间的延长,侵袭分支变长变宽,且具有侵袭分支的比例增多。对组装体进行α-平滑肌肌动蛋白(α-SMA)染色,α-SMA为肌成纤维细胞的标志物。结果显示随着培养时间的延长,组装体总α-SMA表达增强,侵袭分支前端带有小脂滴去分化的脂肪细胞高表达α-SMA,绿色荧光标记的MCF-7细胞紧随其后。说明脂肪细胞转化为肌成纤维细胞,进而带动良性肿瘤细胞MCF-7的恶性侵袭。
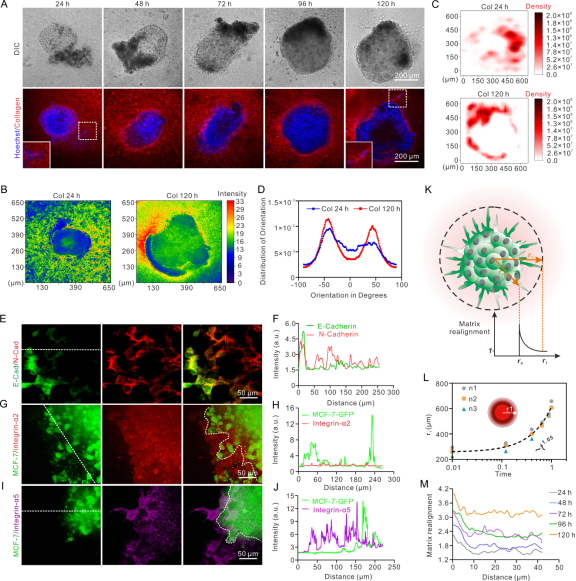
图5展示了脂源性肌成纤维细胞对胶原基质重塑的调控作用。随着培养时间的延长,组装体周围的胶原密度增强,且纤维排列呈现更强的定向性与空间不对称性。去分化的脂肪细胞高度表达N-cadherin并与肿瘤细胞表达的E-cadherin之间形成紧密的异质性细胞连接。整合素α2(一种与胶原蛋白结合的亚型)在脂肪细胞衍生细胞和良性癌细胞中均均匀表达。相反,整合素α5(与RGD基序结合)仅在脂肪细胞衍生细胞中选择性表达。这些结果提示,脂肪细胞转分化为肌成纤维细胞后,通过表达整合素α5与纤连蛋白结合,驱动胶原基质重塑并促进肿瘤细胞迁移。进一步探究基质重塑与肿瘤组织侵袭之间的动力学关系,采用了Ahmadzadeh等人近期开发的理论模型,当细胞在没有外部驱动力的情况时,其移动方式遵循布朗运动规律,其中传播距离(r)会随时间(t)的增加而增大,其指数值为τ=0.5。将其与组装体侵袭的实验数据进行拟合,发现指数值τ达到1.05,显著高于理论值0.5。说明组装体中的细胞迁移并非单纯的随机扩散,而是受到持续的外部驱动力推动。
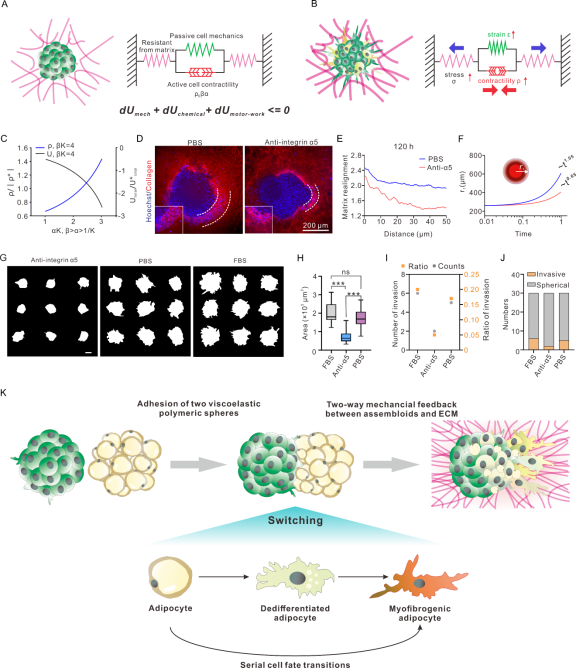
图6描述了肿瘤-脂肪组装体与基质之间的力学互作分析。采用了一个数学模型捕捉了上述两个实体之间的正反馈回路。该模型通过整合与应力纤维组装相关的化学自由能、细胞和ECM变形产生的机械能以及细胞分子马达对ECM所做的机械功来量化细胞的收缩能力和与ECM之间的力学耦合作用。模型的预测结果显示,随着收缩力增加,系统自由能随α的上升趋势而降低,提示优化后的耦合参数α可能会导致细胞收缩力降低,从而降低侵袭性。为验证该假设,使用单克隆抗整合素α5抗体处理肿瘤-脂肪组装体来消除由整合素α5介导的机械反馈后,侵袭距离缩短,基质重排程度降低,τ值从1.05降低到了0.65,表明机械反馈被有效切断,细胞迁移趋向随机扩散,且组装体趋于球形,侵袭面积明显减小,支持其对侵袭行为的抑制作用。
原文链接:
https://authors.elsevier.com/a/1lZlf8YyDfuZU7

