AM J KIDNEY DIS:罕见肾结石疾病的认知与诊疗进展
时间:2025-09-10 12:13:19 热度:37.1℃ 作者:网络
罕见肾结石疾病是一类由单基因突变引起的遗传性肾脏疾病,主要包括原发性高草酸尿症(PH)、胱氨酸尿症、Dent病、腺嘌呤磷酸核糖转移酶(APRT)缺乏症等。这些疾病通常在儿童或青少年期以肾结石或肾钙质沉着症为首发表现,患者常面临肾功能损伤甚至肾衰竭的风险。由于发病率低且临床表现多样,这类疾病的诊断常被延误,而有效治疗手段的缺乏也进一步降低了临床诊断的紧迫性。然而,随着基因检测技术的普及和成本下降,以及对疾病分子机制认识的深入,尤其是新型治疗药物的出现,早期准确诊断和针对性治疗显得尤为重要,有望改变疾病自然进程、保护肾功能并改善患者预后。

本文系统回顾了多种罕见肾结石疾病的病理生理机制、临床表现、诊断策略及治疗进展。原发性高草酸尿症(PH)是由于肝脏中乙醛酸代谢异常导致草酸过度生成,进而引起高草酸尿症、肾结石、系统性草酸盐沉积和进行性肾衰竭。PH分为三种亚型,分别由AGXT、GRHPR和HOGA1基因突变引起。诊断依赖于尿和血浆草酸水平检测,并经基因检测确认。治疗方面,除传统的水化疗法和枸橼酸盐抑制结晶外,针对PH1的RNA干扰疗法(如Lumasiran和Nedosiran)已获批准,能显著降低尿草酸水平,延缓肾功能恶化。对于终末期肾病患者,以往多建议肝肾联合移植,如今在RNAi治疗保驾下可考虑单独肾移植。
胱氨酸尿症是一种常染色体隐性遗传病,由SLC3A1或SLC7A9基因突变导致近端肾小管对胱氨酸等二碱基氨基酸重吸收障碍,尿中胱氨酸过饱和形成结石。其特征性表现为尿中出现六边形胱氨酸晶体。治疗核心是大量饮水、碱化尿液(目标pH 7-8)及采用硫醇结合药物(如噻普罗宁或D-青霉胺)以增加胱氨酸溶解度。Dent病是一种X连锁遗传性肾小管病,主要由CLCN5或OCRL1基因突变引起,表现为低分子量蛋白尿、高钙尿症、肾钙质沉着症和慢性肾脏病。男性患者多见,女性携带者亦可出现轻度症状。治疗重点在于控制高钙尿,采用低盐饮食、噻嗪类利尿剂,并监测肾功能变化。

图1 尿中的胱氨酸晶体
APRT缺乏症则因腺嘌呤代谢障碍导致2,8-二羟基腺嘌呤(DHA)在尿中结晶,形成放射性可透性结石,可引起急性肾损伤和慢性肾病。特征性表现为尿中出现圆形棕色DHA晶体,婴幼儿可见尿布棕红色染色。治疗主要依赖别嘌呤醇或非布司他抑制黄嘌呤脱氢酶,减少DHA生成,配合低嘌呤饮食和充分水化。此外,文章还讨论了与高钙尿和肾钙质沉着相关的遗传性低磷血症性佝偻病(如SLC34A3突变)、CYP24A1突变导致的维生素D代谢异常、家族性低镁血症伴高钙尿和肾钙质沉着症(claudin 16/19突变),以及尿酸代谢异常相关疾病(如Lesch-Nyhan综合征、黄嘌呤尿症和肾小管酸中毒)的临床特点和治疗原则。
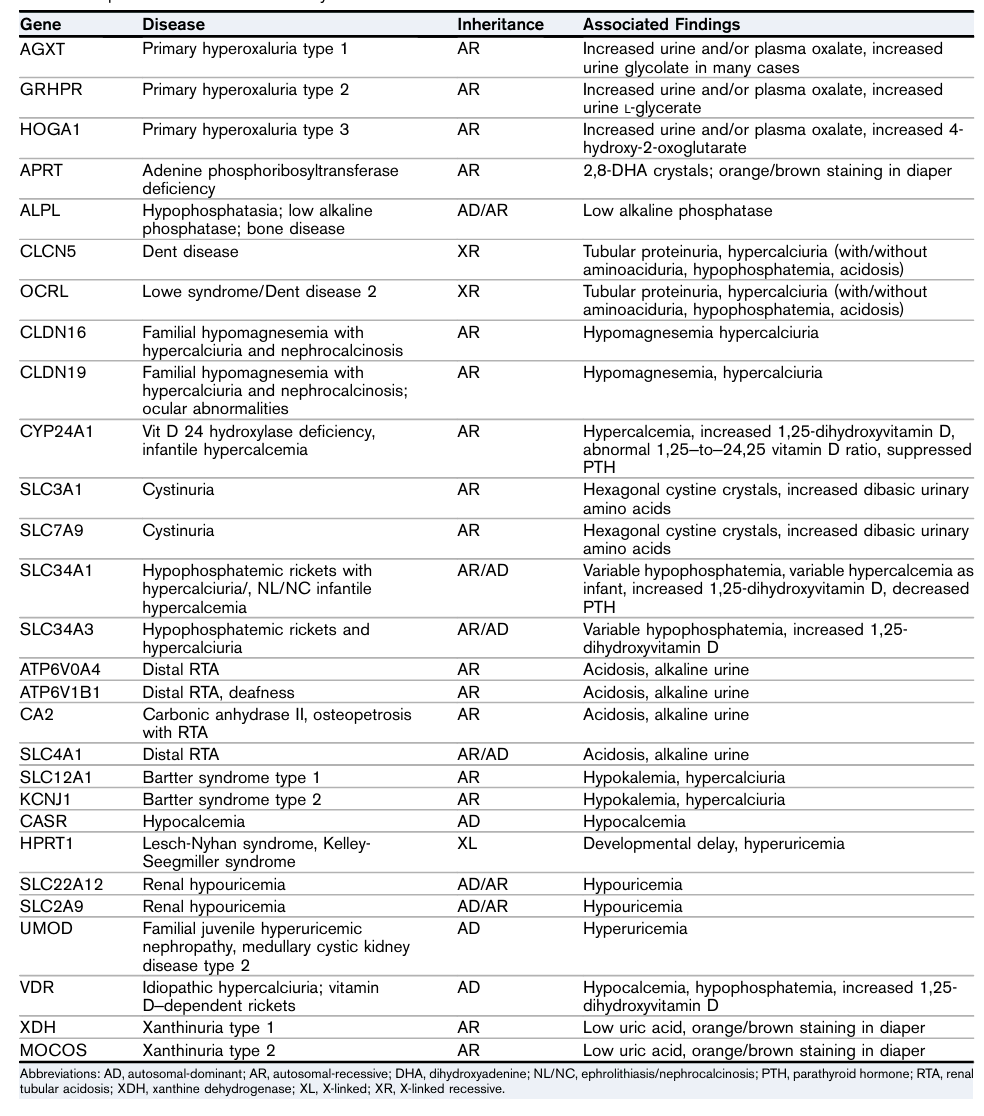
表1 罕见肾结石疾病中的重要基因
诊断方面,文章强调对具有可疑临床特征(如婴幼儿期结石、家族史、复发性结石、不明原因肾衰竭等)的患者应积极进行基因检测。除针对性基因检测外,采用广谱肾结石和肾钙质沉着症相关基因panel检测可能提高诊断率。治疗需多学科协作,包括肾病学家、泌尿科医生和营养师等,个体化制定水化、饮食调整、药物和外科干预策略。对于复杂病例,建议转诊至具有罕见病诊疗经验的医疗中心。患者和家属还可借助疾病特异性倡导组织获取最新研究信息和专家资源。
总体而言,随着对罕见肾结石疾病分子机制的深入理解和新型治疗方法的出现,临床医生应提高对这些疾病的认识和警惕性,尽早通过基因检测明确诊断,并采取针对性治疗措施,以改善患者长期预后和生活质量。
原始出处:
Baum, M. A., Mandel, M., & Somers, M. J. G. (2025). Understanding Rare Kidney Stone Diseases: A Review. American Journal of Kidney Diseases, 86(2), 236–244. https://doi.org/10.1053/j.ajkd.2025.03.023
本文相关学术信息由梅斯医学提供,基于自主研发的人工智能学术机器人完成翻译后邀请临床医师进行再次校对。如有内容上的不准确请留言给我们。

