“腊肠体”:遗传性压力易感性周围神经病(HNPP)|疑难探究
时间:2025-09-13 12:13:39 热度:37.1℃ 作者:网络
论坛导读:遗传性压力易感性周围神经病(HNPP)是一种遗传性运动感觉性神经病,呈常染色体显性遗传,主要症状表现为与压力相关的反复发作性肢体麻木无力。HNPP的神经病理特征性改变为髓鞘增厚形成“腊肠体”样结构,故曾被称为腊肠体样周围神经病。HNPP与外周髓鞘蛋白22(peripheral myelin protein 22,PMP22)表达减少有关,在各年龄阶段均可发病,首发症状多出现在20~30岁。
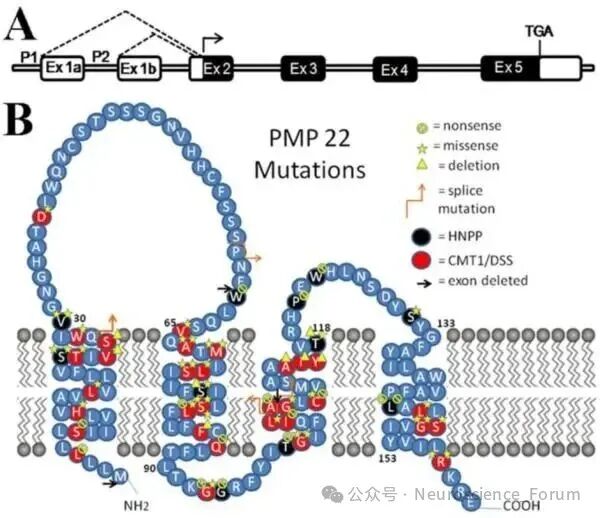
遗传性压力易感性周围神经病(hereditary neuropathy with liability to pressure palsies,HNPP)是由人类周围髓鞘蛋白22(peripheral myelin protein 22,PMP22)基因缺失或点突变引起的一种少见的常染色体显性遗传性周围神经病。HNPP由De Jong于1947年首先提出,患者多在跪着刨土豆后反复出现腓总神经麻痹症状,因此又称为“刨土豆病”。HNPP的发病率为7.3/10 000~16.0/10 000,多于20~30岁起病。HNPP是一种常染色体显性遗传病,多数患者在染色体17p11.2-p12位点存在包含PMP22基因的1.5 Mb的大片段缺失,15%~20%的患者由PMP22基因点突变所致。
HNPP的主要临床表现为肢体麻木或无力,少数患者可出现疼痛、肢体畸形、跛行等。除了典型的反复性压迫性麻痹外,HNPP还存在多种不典型临床表现,常导致误诊或诊断延迟。部分患者表现为缓慢进展的对称性多发性神经病,与Charcot-Marie-Tooth病相似,但没有典型的急性发作史。少数患者始终表现为同一神经的反复受累,如后骨间神经损害导致的抬指不能。约20-30%的基因携带者从未出现临床症状,但神经电生理检查可发现异常,提示为亚临床型HNPP。极少数病例报道有白质异常和偏头痛、抑郁等非典型症状,但其与PMP22缺失的关联尚不明确。
一项临床研究发现 60.7%患者有诱发因素,39.3%患者无诱发因素,这可能与部分HNPP患者起病隐匿有关,或者HNPP存在其他不易察觉的诱发因素。在有诱发因素的患者中,47.1%为肢体牵拉或受压,35.3%为剧烈运动或劳累,还有17.6%为受凉。因此HNPP的诱发因素可能并不仅以肢体受压或牵拉为主,需进一步扩大样本量进行验证。
HNPP电生理主要特点为多发性周围神经运动和感觉功能受损:感觉神经传导速度(sensory nerve conduction velocity, SNCV)明显减慢,可有嵌压部位的感觉神经动作电位波幅减低或消失,运动神经传导速度(motor nerve conduction velocity, MNCV)也可有轻到中度减慢,运动传导主要表现为远端潜伏期延长。约80%的患者存在F波出现率下降或潜伏期延长,反映近端神经段的受累。电生理检查不仅有助于HNPP的诊断,还能评估病变的分布范围和严重程度,为预后判断提供参考。值得注意的是,电生理异常的范围常远超过临床症状,提示HNPP实际上是一种全身性周围神经病变,而不仅仅是局灶性病变。
神经超声检查被证明在周围神经病变的诊断过程中可以对电生理结果起到有效补充。HNPP患者通常在超声上表现为周围神经CSA多灶性增大,尤其是出现在常见的卡压部位。神经MRI也可以对HNPP的诊断起到帮助。HNPP患者的神经MRI显示尺神经和腓总神经的不对称性肿胀及信号增高及神经口径增粗等。
HNPP临床异质性大、相当一部分患者无家族史、或是首次发作就诊,神经电生理检查作为一种快速、准确的检查方法,能发现广泛的亚临床神经损害,对明确该病的诊断极为关键。当患者临床表现为轻微受压后即出现神经麻痹时,应常规行神经电生理检查,并扩大电生理的检查范围。当存在亚临床神经病变时,应高度怀疑HNPP,同时追溯患者家族史,并对患者家属进行神经电生理的筛查,必要时行相关基因检测以提高疾病诊断率。基因检测是确诊HNPP的金标准。
HNPP的诊断标准(2000年Dubourg等):
① 常染色体显性家族史(17p11.2上含PMP22基因、长约1.5Mb的大片段缺失或PMP22基因点突变);
② 发病年龄在20岁左右;
③ 反复发作单神经或多神经麻痹,在轻微损伤时有突然的感觉或运动缺陷,多无痛;
④ 神经电生理检查:正中神经的远端潜伏期增加,传导速度减慢;或其中一侧腓总神经的运动传导速度降低等。
符合上述条件者可考虑诊断为HNPP。临床需要注意与下列的多种周围神经病进行鉴别:
① 腓骨肌萎缩症1A型(CMT1A):与HNPP涉及相同的基因区域(17p11.2),但CMT1A为PMP22基因重复而非缺失。CMT1A表现为缓慢进展的对称性周围神经病,无急性发作特点,电生理检查显示均匀一致的神经传导速度显著减慢(通常<25m/s)。
②慢性炎性脱髓鞘性多发性神经病(CIDP):表现为获得性、进展性或复发性多发性神经病,电生理可见多灶性脱髓鞘改变,但无遗传性基础,脑脊液蛋白-细胞分离,对免疫治疗反应良好。其中要注意变异型之一多灶性获得性脱髓鞘性感觉运动神经病(又称Lewis-Sumner综合征),主要表现为不对称的肢体远端肌肉无力、萎缩、感觉异常,神经电生理表现为局灶性神经髓鞘损害,易与非典型的HNPP相混淆。家族史、诱发因素、脑脊液检查、基因检测等有助于鉴别。
③ 嵌压性神经病:普通嵌压性神经病(如腕管综合征、肘管综合征)通常有明确的局部受压因素,无阳性家族史,电生理检查异常局限于特定嵌压部位,无弥漫性改变。
④ 遗传性神经痛性肌萎缩:表现为反复发作的臂丛神经炎,常有剧烈疼痛,与HNPP的无痛性麻痹不同,且电生理检查缺乏弥漫性异常。
⑤多灶性运动神经病(multifocal motor neuropathy,MMN):以多发性单神经病为主要表现形式,症状主要表现为进行性非对称性肢体无力,上下肢均可受累,上肢重于下肢,远端重于近端。其神经电生理检查可见传导阻滞。易与HNPP混淆。MMN呈慢性病程,传导阻滞存在于神经非嵌压部位,血清抗神经节苷脂GM1抗体检测等有助于鉴别。
目前对HNPP尚无特殊的治疗方法,仍以对症治疗为主。如足下垂可使用踝足矫形器,腕管综合征可使用手腕夹板,疼痛可应用止痛药物等。同时需注意防止长时间的活动和姿势对周围神经的压迫或拉伸损伤。减压手术作用尚不明确,对于部分症状重且长期不缓解的患者可能有效。HNPP预后良好,生存期不受影响,神经功能障碍多在6个月以内自行恢复,少数患者会遗留永久性功能障碍,预后不良与长期局灶神经压迫有关。
HNPP是一种独特且常常被低估的遗传性周围神经病,其临床谱系比传统认识的更为广泛。典型的复发性、无痛性单神经病仍是其主要表现,但不典型表现如慢性多发性神经病和孤立性单神经病并不罕见,这增加了诊断的复杂性。电生理检查在HNPP的诊断中具有核心地位,其特征性的弥漫性传导异常,尤其是远端运动潜伏期延长,往往能提供重要的诊断线索。基因检测(特别是MLPA对PMP22缺失的检测)是确诊的金标准,对于临床或电生理怀疑HNPP的患者应尽早进行。HNPP具有自愈性,早期、精准的诊断可避免不必要的治疗,减轻患者的经济负担和心理压力。
参考文献:
1.Jonathan Morena , Anirudh Gupta, J Chad Hoyle. Treating PMP22 gene duplication-related Charcot-Marie-Tooth disease: the past, the present and the future[J]. Int J Mol Sci . 2019 Jul 12;20(14):3419.
2.Luigetti M, Del Grande A, Conte A, Lo Monaco M, Bisogni G, Romano A, et al. Clinical, neurophysiological and pathological findings of hnpp patients with 17p12 deletion: A single-centre experience. J Neurol Sci. 2014;341:46-50
3.Dubourg O, Mouton P, Brice A, LeGuern E, Bouche P. Guidelines for diagnosis of hereditary neuropathy with liability to pressure palsies. Neuromuscul Disord. 2000;10:206-208
4.de Oliveira AP, Pereira RC, Onofre PT, Marques VD, de Andrade GB, Barreira AA, et al. Clinical and neurophysiological features of the hereditary neuropathy with liability to pressure palsy due to the 17p11.2 deletion. Arq Neuropsiquiatr. 2016;74:99-105
5.Farrar MA, Park SB, Krishnan AV, Kiernan MC, Lin CS. Axonal dysfunction, dysmyelination, and conduction failure in hereditary neuropathy with liability to pressure palsies. Muscle Nerve. 2014;49:858-865
6.Suzan Boutary , Andoni Echaniz-Laguna, David Adams, Julien Loisel-Duwattez , Michael Schumacher, Charbel Massaad, Liliane Massaad-Massade. Treating PMP22 gene duplication-related Charcot-Marie-Tooth disease: the past, the present and the future[J].Transl Res.2021Jan;227:100-111.

