头颈部癌中临床相关基因融合的发生率,潜在获益的靶向治疗
时间:2025-09-12 12:11:06 热度:37.1℃ 作者:网络
头颈部癌(HNC)是全球第七大常见癌症。目前获批的全身治疗包括化疗、抗EGFR抗体和PD-1免疫治疗,基于基因组的靶向治疗较少。在其他实体瘤中,涉及FGFR、NTRK、ALK等致癌激酶基因的融合具有临床可靶向性,但它们在头颈部癌中的发生率相关知识有限。本研究在超过13,000例头颈部癌肿瘤(不包括唾液腺肿瘤)的合并数据集中,描述了致癌融合的基因组谱及其生物学影响。研究者发现66例(2.8%)存在致癌融合,包括既往报道的FGFR3融合(n=19)和功能获得性EGFR融合(n=6)。融合阳性头颈部癌的基因表达显著更高,人乳头瘤病毒(HPV)感染率显著高于融合阴性头颈部癌(p<0.001)。与野生型相比,存在FGFR变异的肿瘤细胞增殖更活跃,NK细胞和CD8+T细胞数量更多。本研究结果为头颈部癌患者提供了更多治疗机会。
研究背景
头颈部癌(HNC)是全球第七大常见癌症,5年生存率约为50%。这种侵袭性疾病可发生于多个解剖部位,包括口咽、口腔、喉、鼻窦、唾液腺和鼻腔。在头颈部癌的发生中起重要作用的环境因素包括烟草、酒精和人乳头瘤病毒(HPV,主要是HPV16)感染。这些因素通常相互排斥。由于头颈部癌的异质性和常被晚期诊断,其治疗仍面临挑战。目前获批的全身治疗包括放疗,联合顺铂化疗或抗EGFR抗体(西妥昔单抗),作为一线治疗或术后治疗。然而,缓解率仍然有限,在13%至45%之间。PD-1免疫治疗也已成为复发和转移性头颈部癌患者的另一种治疗选择,但总缓解率仍维持在16%-20%。一项研究显示,PD-1抑制剂纳武利尤单抗联合西妥昔单抗可将1年总生存率提高至66%,中位生存期达20.2个月。另一项研究报道,帕博利珠单抗联合西妥昔单抗试验的6个月总缓解率为45%,还有一项研究发现西妥昔单抗联合度伐利尤单抗试验的缓解率为39%,中位缓解持续时间为8.6个月。然而,抗EGFR抗体与免疫治疗的联合疗法用于晚期头颈部癌患者时,并未进行筛选,不考虑EGFR突变或PD-L1免疫组织化学状态。因此,需要在头颈部癌中确定更多基因组生物标志物,以扩大靶向治疗的适用人群,并为早期患者提供个性化治疗选择。
虽然头颈部癌的肿瘤基因组变异已有描述,但基于分子亚群的个性化治疗在该疾病中并未取得显著进展。NGS加深了我们对头颈部癌的理解。这些肿瘤常存在抑癌基因(如NOTCH1、PTEN)的变异,以及致癌通路(包括JAK/STAT通路)的异常。此外,在头颈部癌中已观察到具有临床可干预性的FGFR3-TACC3基因融合,以及癌基因(包括PIK3CA、FGFR1和EGFR)的拷贝数变异。尽管如此,针对头颈部癌患者的靶向治疗成功开发的较少,部分原因是人们认为不存在具有临床可干预性的靶点。目前,只有携带HRAS点突变(3%-4%)的患者符合使用替吡法尼进行个性化治疗的条件。这与其他癌症类型形成对比,在其他癌症中,精准肿瘤学方法应用更广泛,例如肺腺癌和结直肠癌,分子靶点可指导治疗选择。本研究旨在确定头颈部癌中可能适合个性化靶向治疗的基因组变异。
基因融合在多种不同癌症类型中具有临床可干预性,但在头颈部癌中尚未得到充分研究。许多涉及激酶基因的融合,包括ALK、ROS1、FGFR1、FGFR2、FGFR3、RET、NTRK1、NTRK2和NTRK3,可通过临床获批的TKIs进行靶向治疗。其他涉及EGFR、BRAF和NRG1的致癌融合显示出用酪氨酸激酶抑制剂进行靶向治疗的潜力。与拷贝数变异和点突变等其他变异类型相比,头颈部癌中基因融合的描述较少。案例研究和小队列研究中已观察到涉及癌症增殖或基因转录相关基因的融合,如BRAF、ROS1、FGFR和CD274。尽管有这些报道,但对头颈部癌中临床相关基因融合及其生物学影响的了解仍然有限,原因有二:(1)头颈部癌研究样本量小,通常聚焦于单一解剖部位,因此不足以全面分析罕见变异(如基因融合);(2)少数利用大数据(n>200样本)的研究采用了现已过时的生物信息学方法进行基因融合分析。例如,TCGA使用MapSplice检测基因融合,发表了其多组学、全面的头颈部鳞状细胞癌数据集(n=279患者)。利用这种用于识别剪接变异的方法,他们发现了2例融合阳性病例,均携带FGFR3-TACC3融合。Stransky等人使用更新后的TCGA队列(源自原始研究,共411例头颈部癌患者),对激酶融合进行了RNA测序分析。他们发现了与TCGA研究者描述的相同的2例FGFR3-TACC3融合阳性病例,以及另外2例融合阳性病例,1例携带NTRK2融合,另1例携带NTRK3融合。自这些研究发表以来,已开发并优化了多个基因融合软件包,以提高基因融合检测效果。在一项比较各种基因融合软件的研究中,结果表明STAR-Fusion和Arriba在平衡敏感性和特异性方面是最佳的基因融合工具之一。事实上,在该研究发表后,研究者利用这些工具重新分析了整个TCGA数据集的基因融合,目前该结果可在GDC门户网站获取。然而,仍需要明确哪些基因融合代表功能获得性事件,且可能成为治疗靶点。本研究通过对最大规模的头颈部癌队列(n=13,655例肿瘤)进行研究来解决这一问题,旨在描述功能获得性融合的发生率、其生物学影响,并确定携带EGFR和FGFR反复变异的头颈部癌患者是否对酪氨酸激酶抑制剂治疗敏感。
为发现并明确可能从基因融合靶向治疗中获益的头颈部癌患者队列,研究者利用了配对RNA测序、全外显子测序和临床数据的数据集。研究者整合多个数据源,构建了包含超过13,000例所有解剖部位(不包括唾液腺)和所有组织学类型的头颈部癌病例队列,以识别并描述头颈部癌中致癌基因融合的发生率及其生物学和临床影响。研究者证实,具有临床相关性基因融合的肿瘤在头颈部癌中存在一定发生率,其基因表达更高,HPV阳性率更高,并表现出独特的免疫表型。这些发现可为携带基因融合的头颈部癌拓展个性化治疗选择。
研究结果
基因融合存在于头颈部癌的多个解剖部位:
研究者整合多个数据源,构建了包含2564例的研究队列和11,091例的临床数据库,共13,655例头颈部癌病例。这些数据源包括ORIEN(n=1441)、TCGA(n=528)、CPTAC(n=110)以及来自dbGaP和ArrayExpress的17个已发表数据集(n=485)。采用STAR-Fusion和Arriba软件分析这些病例的RNA测序数据以检测基因融合。通过该方法,研究者识别出66例(发生率2.57%)携带具有临床相关基因伴侣的基因融合(图1A)。大多数融合阳性病例为鳞状细胞癌(64/66),1例为腺癌,1例为腺鳞癌(图1B)。研究者发现涉及已证实具有临床可靶向性的致癌基因的融合,包括FGFR3(n=19)、EGFR(n=6)、FGFR2(n=6)和NRG1(n=5)(图2A)。基因融合最常见的解剖部位是口咽(n=25),其次是口腔(n=20)和喉(n=17)(图2B)。利用OncoKB数据库,研究者发现66例基因融合中48例(72.7%)被归类为“致癌性”或“可能致癌性”。此外,通过分析Caris Life Sciences临床数据集中11,091份样本的RNA测序数据,研究者观察到54个临床相关基因融合,包括涉及FGFR3(n=43)、FGFR2(n=2)和EGFR(ERBB1)(n=2)的变异。值得注意的是,该临床数据集对报告既往已发现的基因融合有严格标准。尽管如此,其整体结果与发现队列相似(图2),基因融合最常见的解剖部位仍是口咽。
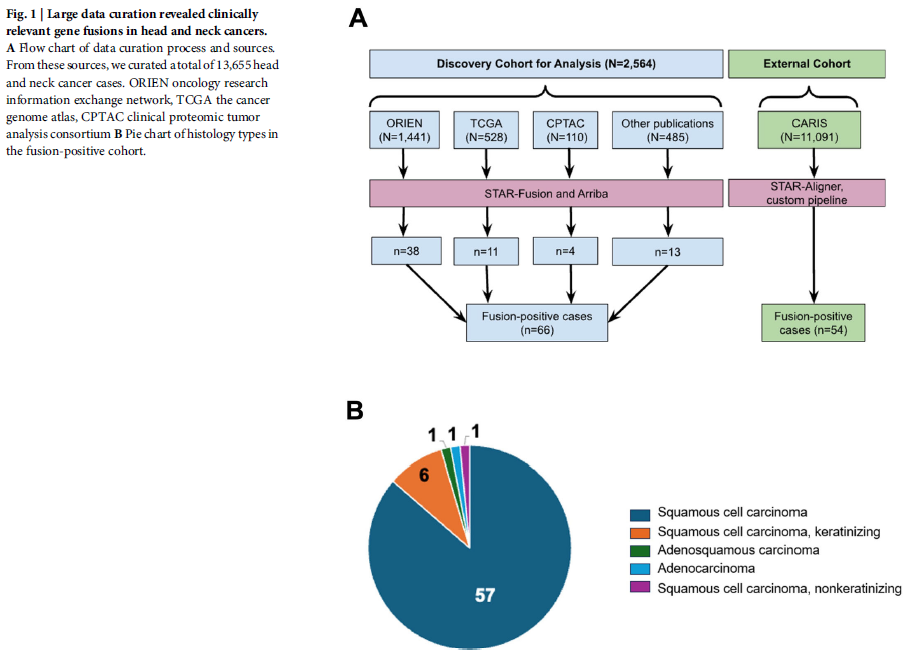
图1
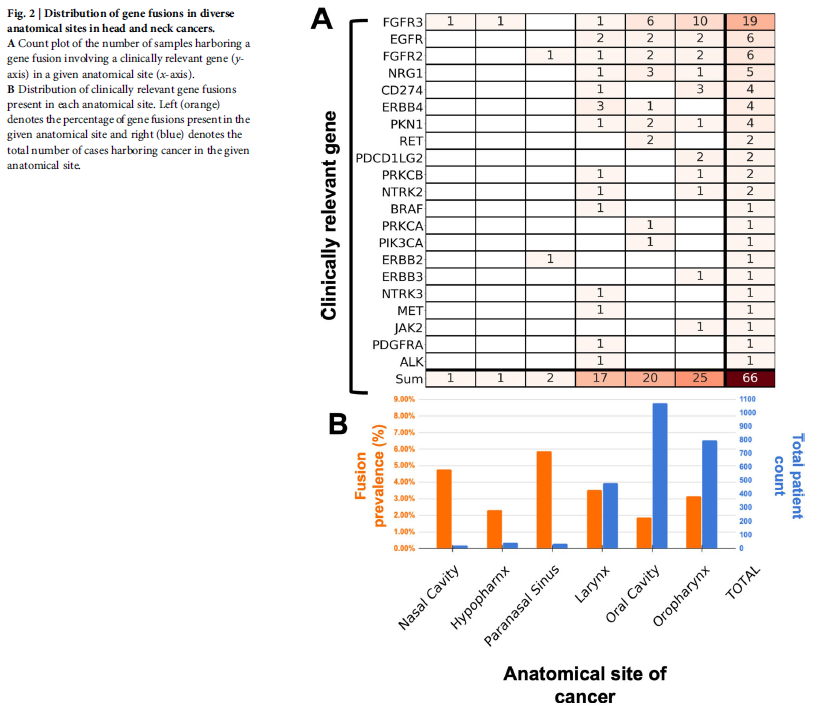
图2
头颈部癌中存在功能获得性EGFR融合:
令人意外的是,研究者在合并队列中识别出6例EGFR融合。尽管本研究聚焦于鳞状细胞癌,但在1例唾液腺癌患者(ORIEN队列)中也观察到EGFR融合(该癌种中转录因子和NTRK融合更为人熟知)。研究者发现与EGFR相关的多种融合伴侣(图3),其中大多数以EGFR为5'端伴侣(n=5)(图3A)。有趣的是,研究者还观察到以EGFR为3'端伴侣的融合,其激酶结构域保持完整(n=2)(图3B)。其中4例病例携带两个特定断点之一,这些断点既往已被证实具有功能获得性,且/或在接受EGFR TKI治疗的患者中显示出临床应答。通过对EGFR融合相关文献的评估,17例接受EGFR TKI治疗的患者中,15例部分缓解,1例完全缓解,1例疾病稳定。这些病例中大多数为肺腺癌(n=16/17),1例为结直肠腺癌(n=1/17)。与肺腺癌中常见的EGFR突变一致,近半数融合病例(8/17)为非吸烟者。最常见的EGFR融合断点位于外显子24(包含外显子1-24),即激酶结构域末端,此时EGFR为5'端伴侣(n=8/17)。研究者还观察到断点位于外显子18(包含外显子18-28)(即激酶结构域起始处)的基因融合(n=2/17)。将这些病例与本研究的EGFR融合阳性队列对比,发现4/6例病例携带与应答者中观察到的相同EGFR融合结构:3例EGFR断点位于外显子24(5'端伴侣),1例断点位于外显子18(3'端伴侣)。从融合数据分析和整理的文献中可见,最常见的EGFR断点位于外显子24末端,该外显子是编码激酶结构域的最后一个外显子。基于此,研究者利用来自TCGA(n=10,533)和ORIEN(n=11,489)的泛癌队列,发现12种独特癌症类型中共有66例存在EGFR外显子24断点融合。
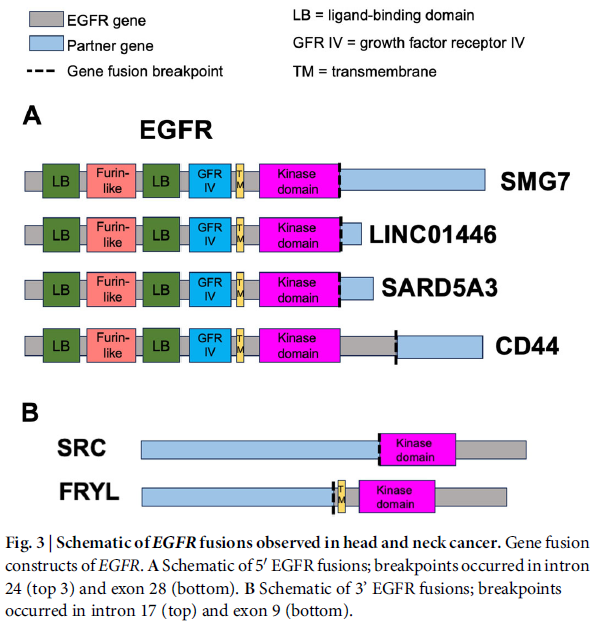
图3
临床相关基因融合表现出转录本表达上调:
接下来,研究者分析了融合阳性头颈部癌的生物学影响。利用来自两个大型队列(TCGA,n=528样本;ORIEN,n=1618样本)以及合并的488份来自文献的整理RNA测序样本的RNAseq数据,研究者发现融合阳性病例中其各自临床相关基因融合伴侣的表达上调。为分析基因融合阳性癌症与基因表达上调的关联,研究者通过将计数转换为z分数对RNA计数进行标准化。结果发现,在上述每个数据集中,融合阳性病例中其特定基因伴侣的z分数显著高于融合阴性病例(p<0.001)(图4A-C)。
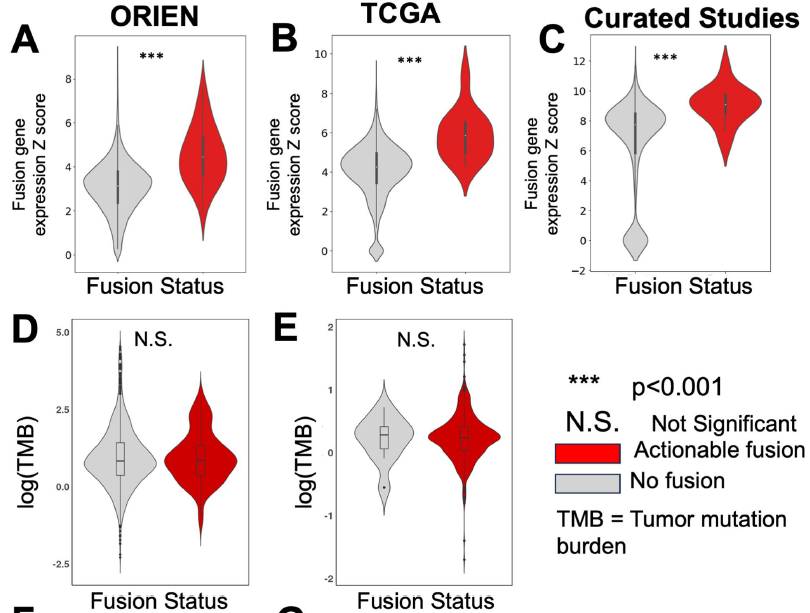
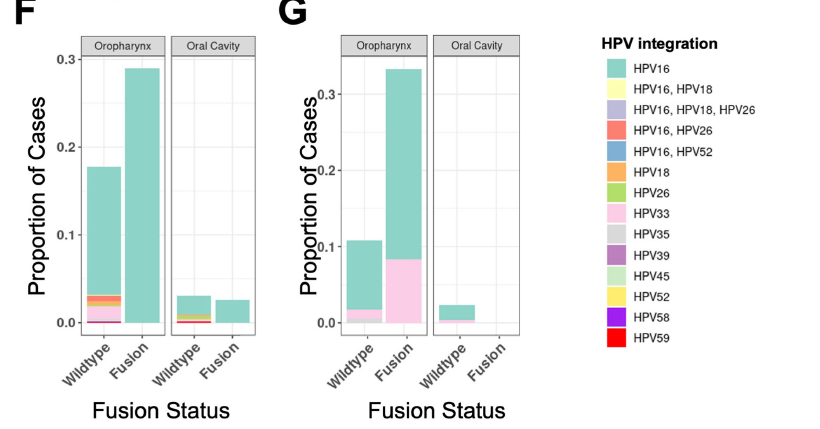

图4
临床相关基因融合无独特的共突变模式:
由于致癌基因融合与低肿瘤突变负荷相关,研究者研究了这些肿瘤的突变状态。在ORIEN的42例融合阳性样本中,31例有可用于突变分析的全外显子测序数据。值得注意的是,将ORIEN中的融合阳性队列(n=31)与融合阴性组(n=1579)对比,发现肿瘤突变负荷无显著差异(p>0.5)(图4D)。对TCGA中融合阳性样本(n=12)与融合阴性样本(n=516)的肿瘤突变负荷分析也得出一致结果(图4E)。此外,研究者研究了与融合阳性病例相关的致癌共突变。在ORIEN中,31例融合阳性病例中有6例携带PIK3CA(n=4)、NOTCH1(n=1)、HRAS(n=1)和CDK2NA(n=1)的功能突变,其中1例同时携带PIK3CA和HRAS突变。在TCGA中,12例病例中有3例携带PIK3CA(n=2)和TP53(n=1)的致癌突变。值得注意的是,这些融合阳性病例与FGFR2、FGFR3、PTEN和NRAS的功能突变互斥。
临床相关基因融合在HPV阳性头颈部癌中更常见:
由于HPV与部分头颈部癌相关,研究者评估了融合阳性病例中HPV的发生率。总体而言,在ORIEN和TCGA中,融合阳性队列中HPV阳性样本的比例高于融合阴性队列(图4F、G)。在ORIEN队列中,18%(250/1376)的融合阴性患者为HPV16阳性,而融合阳性队列中HPV16阳性率更高,为31%(12/37)(p=0.05,比值比(OR)=2.0)(图4F)。这些病例主要来自口咽肿瘤(n=11),其次是口腔(n=1),分别占融合阳性队列的28.9%和2.6%(图4F)。值得注意的是,在15例融合阳性口咽肿瘤中,73%(n=11/15)为HPV16阳性,而融合阴性肿瘤的HPV16阳性率略低,为62%(n=244/391)。特别是,研究者观察到FGFR3融合阳性患者中HPV16显著富集(p=0.015,OR=5.44)。研究者在头颈部肿瘤的RNA样本中识别出10种不同的HPV亚型,但在融合阳性组中,仅观察到HPV16(n=12)和HPV18(n=1)。同样,在TCGA中,42%(5/12)的融合阳性病例为HPV阳性,而野生型组为14%(74/509)(p=0.024,OR=4.19)。与ORIEN队列的结果一致,这些病例主要来自口咽肿瘤(n=3),其次是口腔(n=1)和下咽(n=1),分别占融合阳性队列的25%、8.3%和8.3%(图4G)。研究者还注意到,2例FGFR3融合阳性病例中均存在HPV16,与ORIEN数据集中观察到的模式相似。
FGFR变异头颈部癌患者对FGFR TKI治疗应答良好:
由于FGFR融合在本队列中发生率最高,研究者进一步研究了该亚组头颈部癌,包括融合以及功能获得性点突变。尽管队列中最常见的融合是FGFR3-TACC3(n=15),但研究者还观察到其他具有相同断点但不同融合基因伴侣的FGFR3融合(n=4)(图5A)。此外,研究者识别出6例涉及FGFR2的融合,涉及5个独特的融合伴侣(图5B)。其中5例病例的FGFR2断点与FGFR2融合的临床病例中观察到的断点一致,且这些临床病例对FGFR激酶抑制剂治疗有应答。为寻找FGFR1/2/3的其他激活突变,研究者利用ORIEN(n=1610)、TCGA(n=528)和CPTAC(n=110)的全外显子数据。研究者从ORIEN中识别出17例FGFR3点突变,从TCGA中识别出11例,共24个独特肿瘤中观察到28个突变。这些突变事件与基因融合阳性病例互斥。如预期,发生率最高的点突变是FGFR3 S249C(n=16)。研究者观察到的其他功能获得性突变包括FGFR3 G380R(n=4)、R248C(n=4)、R669G(n=2)、V50I(n=1)和D786N(n=1)(图5C)。在CPTAC队列中未发现功能获得性点突变(n=0/110),且在所有数据集中均未发现FGFR1或FGFR2的突变。总体而言,在本研究的合并队列中,共识别出49例携带FGFR2或FGFR3变异的病例,发生率为1.84%(n=49/2564)。
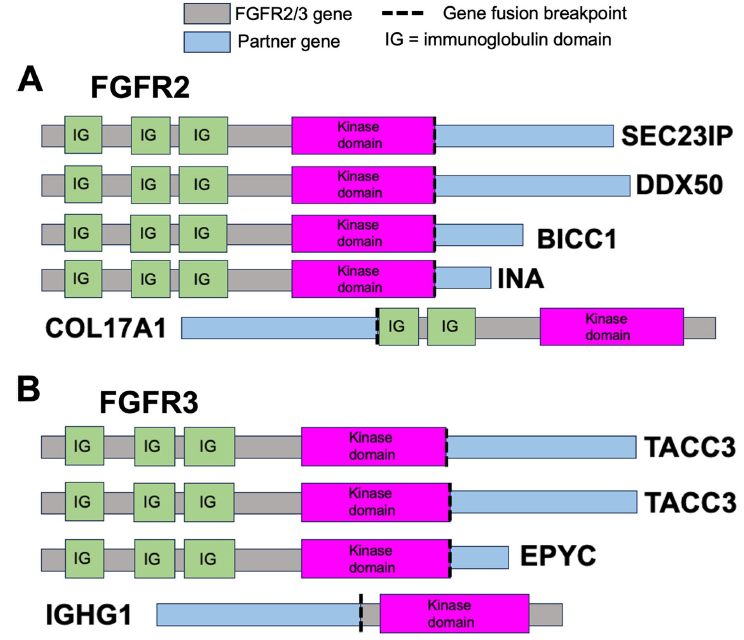
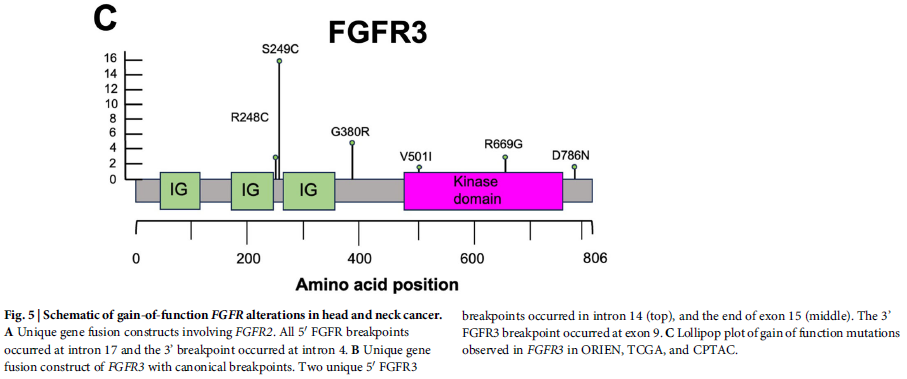
图5
鉴于FGFR激酶抑制剂在其他携带FGFR变异的癌症类型(胆管癌和膀胱癌)中取得成功,研究者旨在确定FGFR变异的头颈部癌对FGFR抑制剂治疗的应答情况。研究者对文献进行全面回顾,以寻找携带FGFR变异的头颈部癌病例接受FGFR抑制剂治疗的报告和既往临床试验。研究者从8项独立研究中发现19例病例对FGFR抑制剂治疗有持久应答(表1)。总体而言,11/19例(58%)至少达到部分缓解,其中2例完全缓解。平均治疗持续时间为10个月。所使用的治疗药物包括FGFR选择性激酶抑制剂福巴替尼、厄达替尼和佩米替尼,以及除FGFR外还靶向多种酪氨酸激酶的药物,包括尼达尼布(nintedanib)、仑伐替尼(lenvatinib)和帕唑帕尼(pazopanib)。
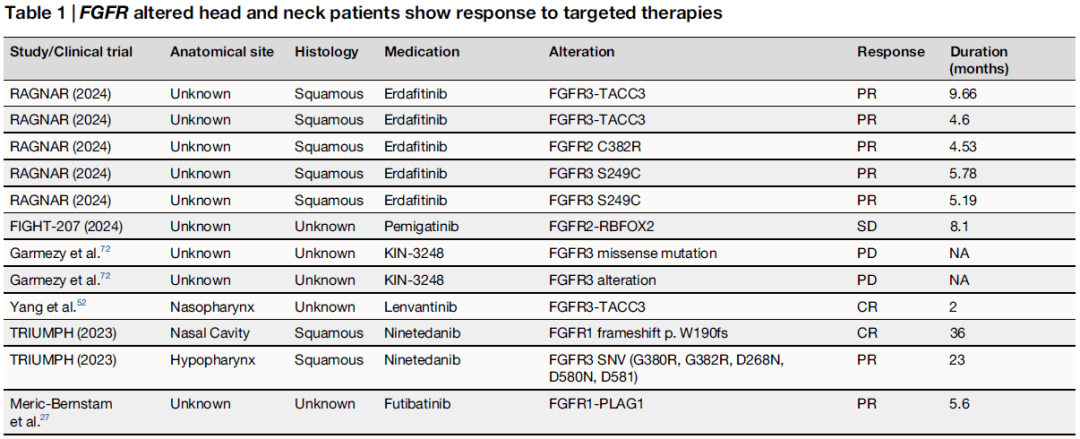

表1
FGFR变异的头颈部癌表现出独特的肿瘤免疫表型:
既往研究表明,FGFR信号传导可改变肿瘤微环境,且有证据显示免疫检查点抑制剂联合FGFR激酶抑制剂治疗可增强FGFR突变癌症的抗肿瘤活性。因此,研究者旨在描述FGFR突变头颈部癌对肿瘤免疫微环境的影响。利用CIBERSORT、Quantiseqr和ImSig分析ORIEN RNAseq数据集中的生物学特征和免疫浸润(该数据集有可用的RNAseq样本)。研究者按突变类型将FGFR变异病例分组:FGFR融合(n=15),包括FGFR2(n=5)和FGFR3(n=10)融合;以及FGFR3点突变(n=12)。尽管FGFR融合阳性组与FGFR野生型组在细胞毒性T细胞(CD8+ T细胞)或CD4激活T细胞数量上无差异,但研究者观察到FGFR3点突变组中这两种细胞类型均显著上调(图6A、B)。此外,与野生型相比,FGFR3突变组中CD4静息T细胞水平更低(图6C)。Quantiseqr分析显示,与野生型相比,FGFR3突变组中自然杀伤(NK)细胞水平更高(图6D)。值得注意的是,研究者观察到ImSig定义的细胞增殖特征在FGFR融合组和FGFR突变组中均显著上调(图6E)。
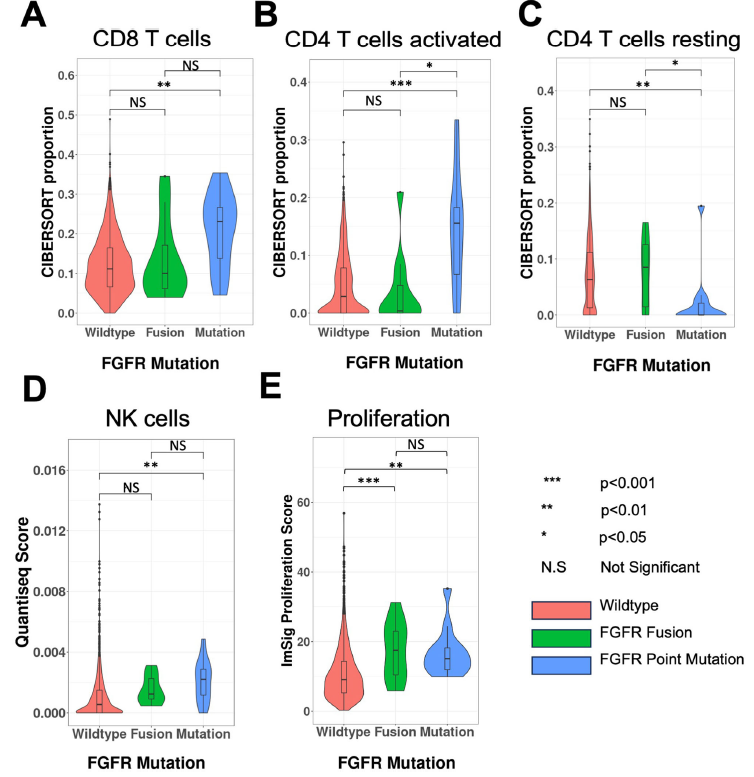

图6
讨 论
本研究描述了HNC中潜在可靶向基因融合的情况。虽然多种癌症类型已有多种已确立的用于靶向治疗的基因组生物标志物,但头颈部鳞状细胞癌仅有一个:HRAS 中的点突变,其仅存在于 3–4% 的 HNC 中。因此,研究者试图发现HNC 中其他可靶向的变异,并扩大潜在的治疗机会。总体而言,研究者鉴定出66个临床相关的基因融合,发生率为 2.73%(66/2564)。研究者在HNC 肿瘤中观察到的这种基因融合发生率与文献中描述的其他实体瘤相当。在实体瘤中,临床相关的基因融合发生率约为 1–2%。具有获批疗法的基因融合,包括 NTRK、RET和 FGFR,在实体瘤中的发生率分别为 0.3%、0.8–1% 和 0.5–0.9%。特定的癌症类型,如结直肠癌、胰腺癌和非小细胞肺癌,其携带的临床可靶向融合发生率约为 1%。此外,本分析发现了先前未在HNC 中描述过的 EGFR 融合。这些 EGFR 融合具有先前在对 EGFR TKI 疗法有反应的肺癌患者中观察到的典型断点。最后,研究者提供的证据表明,携带FGFR 融合的 HNC 患者对 FGFR TKI 抑制剂有反应。总之,这些结果为扩大 HNC 患者的靶向治疗资格提供了证据。先前的研究低估了基因融合在 HNC 中的发生率和影响;然而,研究者表明,利用大型数据集和用于基因融合分析的优化生物信息学工具,揭示了具有生物学影响且比以往认为更常见的变异。
研究者推测,用于基因融合分析的小队列和较旧的生物信息学工具导致了对HNC中临床相关基因融合的了解有限。先前的研究包含不到 100 个病例,这种样本量不足以对基因融合进行全面分析,而基因融合在泛癌中的发生率约为 1%49。迄今为止,关于 HNC 中基因融合的报道仅限于病例研究。已有一些关于 HNC 的更大规模的基因组研究;例如,TCGA 研究人员在 2015 年对 HNC(n = 279 个肿瘤)进行了分析。他们利用当时的现代生物信息学软件 Mapsplice,仅鉴定出 2 种激酶融合,均涉及 FGFR3。Mapsplice 可以通过检测“剪接连接”来鉴定基因融合,本质上是将这种变异视为剪接事件。相比之下,设计用于检测基因融合事件的较新软件,如STAR-Fusion 和 Arriba,同时利用连接读数和不一致的读对,提高了检测额外基因融合的敏感性。不一致的读对发生在配对的读数被映射到不同基因的情况下,这也可能表明存在基因融合事件。结合这两种类型的读数显著提高了算法的性能。Gao 等人使用这些和其他融合检测工具对 TCGA 队列进行了综合融合分析。研究者在TCGA 中观察到的 9 种融合中,有 6 种来自这项先前的研究。在该出版物之后,TCGA 使用 STAR-Fusion 和 Arriba 重新分析了用于基因融合的 RNA 测序数据,从中研究者在TCGA 中观察到 3 种额外的临床相关基因融合,这些尚未被报道。鉴于发现了额外的融合,研究者试图收集更大的HNC 队列。研究者将更新的TCGA分析与来自 ORIEN、CPTAC 和 Caris Life Sciences 的大型数据集以及 17 项已发表研究的数据相结合,创建了一个包含 13,655 名头颈部癌病例的合并队列。据研究者所知,除了唾液腺癌研究外,这是已报道的最大规模的HNC 病例队列。研究者将优化的生物信息学工具STAR-Fusion 和 Arriba 应用于这个大型 RNA 样本数据集,创造了一个最佳环境,以全面鉴定和描述 HNC 中临床可靶向的基因融合。本研究的方法满足了在头颈部癌病例中进行全面基因融合检测所需的更大队列和改进的生物信息学方法这一未被满足的需求。
出乎意料的是,本研究在HNC 中发现了 6 种涉及 EGFR 的新型基因融合。EGFR 融合是一种罕见的变异,发生在0.4% 的癌症病例中,主要在胶质母细胞瘤和非小细胞肺癌(NSCLC)中观察到。这些 HNC 病例中的大多数在 EGFR 基因伴侣中携带断点,这些断点在对 EGFR TKI 治疗有反应的患者中已被观察到。据研究者所知,这是首次报道HNC 中潜在可靶向的 EGFR 基因融合。虽然最近有报道称头颈部癌中存在 EGFR 融合,但这些研究没有区分所观察到的断点或结构的功能获得性融合事件与乘客融合事件。例如,仅保留外显子 1 的反复出现的 EGFR 融合可能没有功能,也不具有临床可干预性。文献整理显示,患有其他携带EGFR 融合的癌症类型的患者对 EGFR TKI 疗法反应良好,包括一例结直肠癌病例,这在该癌症类型中先前未被描述过。所有病例都显示出从 EGFR TKI 治疗中获益。鉴于此,以及本队列中许多EGFR 融合阳性的 HNC 病例携带在这些有反应的患者中观察到的相同的 EGFR 融合断点和结构,研究者认为EGFR 融合阳性的 HNC 病例可能从 EGFR TKI 疗法中获益。6 例 HNC EGFR 融合病例中有 5 例来自 ORIEN 数据库,该数据库包括用药史。正如预期的那样,这些患者中没有(0/5)接受过 EGFR TKI 治疗;然而,令人惊讶的是,他们中没有一个接受过西妥昔单抗这种 EGFR 疗法,而西妥昔单抗是 HNC 的常规治疗方法。研究者推测,可能存在对西妥昔单抗有特殊反应的患者,他们可能存在潜在的EGFR 基因融合。未来,研究者希望研究携带EGFR 融合的 HNC 患者对西妥昔单抗和 EGFR TKI 抑制剂的反应。出于对 HNC 中 EGFR 融合发生率的兴趣,研究者利用泛癌队列,在几种肿瘤类型中发现了额外的EGFR 融合,这些肿瘤类型中 EGFR 融合尚未被报道或描述不充分。这些观察结果表明,除脑癌和肺癌外,其他癌症患者也可能携带具有临床可靶向性的 EGFR 融合。
研究者发现,FGFR变异在本 HNC 队列中的发生率为 1.8%(70/2564 例)。FGFR变异包括基因扩增、点突变和基因融合,是HNC 中最常发生变异的受体酪氨酸激酶基因之一,使其成为FGFR 抑制剂疗法的一个有前景的靶点。在本研究的融合阳性HNC 队列中,FGFR 融合的发生率最高(16/66)(图 1A)。这与文献一致,文献中报道了 HNC 中的 FGFR 融合阳性病例,最常见的是 FGFR3 融合;然而,在临床病例报告中也观察到了 FGFR2 融合。研究者整理了报道接受FGFR TKI 治疗的 FGFR 变异的HNC 患者的临床试验研究和病例研究。重要的是,大多数 FGFR 变异的病例在FGFR TKI 治疗中至少达到部分缓解(表 1),表明这些变异可能成为该疾病的有效预测生物标志物。对FGFR 抑制剂治疗反应最好的患者是一名 38 岁的鼻腔癌女性,她携带 FGFR1 移码缺失;她接受多靶点 TKI 尼达尼布治疗 36 个月,肿瘤缩小 81.4%,达到部分缓解74。本研究结果表明,HNC 中具有临床可干预性的FGFR融合的发生率高于预期。需要更多的工作来确立 FGFR 融合作为 HNC 中的基因组靶点,包括第二代 FGFR 抑制剂进入临床试验。
鉴于在其他癌症(如膀胱癌)中FGFR变异与肿瘤免疫微环境改变相关,研究者进一步表征了这些FGFR变异的HNC。尽管由于样本量小,仅针对 FGFR 融合病例的证据并不确凿,但 FGFR 变异的肿瘤与野生型肿瘤之间存在明显的生物学差异。根据ImSig 的 RNA 特征,FGFR 变异的肿瘤显示出增加的细胞增殖。根据Quantiseqr,FGFR 突变的 HNC 还与静止 CD4+ T 细胞比例较低以及 NK 细胞、CD4+ 活化 T 细胞和 CD8+ T 细胞水平较高相关。NK 细胞在抗肿瘤反应中发挥重要作用。研究表明,PD-1 可能作为 NK 细胞的检查点,PD-1 阻断可增加 NK 细胞活性。CD8+ T 细胞是公认的炎症标志物;CD8+ T 细胞丰度与免疫治疗的无进展生存期呈正相关。有趣的是,FGFR 突变在不同癌症中对免疫微环境的影响似乎不同。与野生型相比,FGFR3 突变的膀胱癌表现出免疫抑制表型,其特征是免疫浸润水平较低和 T 细胞耗竭增加。另一方面,Zhang 等人表明,FGFR 突变的黑色素瘤与较高水平的 CD4+ T 细胞相关,这与本研究发现一致。尽管存在这种不一致,但临床前研究中有证据表明,FGFR TKI 可以增强多种癌症的肿瘤免疫原性,表明在治疗策略中联合 PD-1 免疫疗法和 FGFR TKI 的潜力。研究者推测,具有FGFR 突变的 HNC 患者可能受益于联合治疗方案。研究者承认,该样本量有限(n = 28);因此,需要更大的 FGFR 突变 HNC 队列来进一步了解其独特的相关生物学表型。
尽管本研究鉴定出了临床相关的基因融合,但仍存在一些重要的局限性。研究者关于头颈部癌中基因融合分布的结果来自合并的发现数据集(n = 2564)和 Caris Life Sciences 数据集(n = 11,091)。由于队列的目的不同,其分布也有所差异。合并数据的发现队列的组装和分析旨在揭示头颈部癌中临床相关的基因融合。Caris 的生物信息学流程是为了鉴定和确认已确立的临床可靶向变异的存在而开发的,作为肿瘤诊断报告的一部分;因此,过滤方法更为严格,以确保真阳性结果。
本研究的另一个局限性是队列的抽样偏差。头颈部癌每个解剖部位的治疗方案不同;更常可手术切除的部位的肿瘤,如口腔,比难以接近的部位(包括喉和鼻腔)更频繁地进行基因组测序分析,导致可手术切除的癌症类型被过度代表。一项研究估计,HNC 最常见的解剖部位是口咽(31.1%),其次是口腔(29.1%),然后是喉(28.6%)。相比之下,在本研究的HNC 队列中,最常见的解剖部位是口腔(40.4%),其次是口咽(30.0%)和喉(18.0%),显示出口腔肿瘤被过度代表。显然,需要对所有头颈部肿瘤进行更常规的基因组测序,以全面了解这种癌症类型中可靶向变异的发生率。另一个局限性是所分析的样本标本,特别是来自ORIEN 队列的标本。对于 FGFR 变异的免疫表型分析,研究者利用了RNA 测序数据,其中 15.6% 来自淋巴结。由于淋巴结是免疫细胞的来源,导致免疫浸润水平高于其他样本部位,这可能影响本研究观察到的结果。然而,淋巴结来源的样本在突变组和野生型组之间分布均匀。研究者利用RNA测序数据发现了头颈部癌中新型的可靶向变异和基因融合。DNA 测序可用于分析体细胞突变、拷贝数变异和其他异常,并且在肿瘤基因组分析中比RNA 测序数据应用更广泛。尽管有生物信息学算法可以检测可能包括基因融合的基因组结构变异,但 RNA 测序比 DNA 测序方法更适合基因融合检测,并且基于 DNA 的方法经常会错过 RNA 测序中检测到的基因融合事件。本研究结果表明,RNA 测序是一项重要的基因组分析,应与 DNA 测序方法结合使用,以进行更完整的肿瘤分析,从而检测否则会被遗漏的可靶向变异。
研究者从这项研究中获得的发现为研究头颈部癌中的可靶向变异提供了未来的方向。有必要进一步表征FGFR 变异的头颈部癌中的免疫微环境,以评估包括PD-1 免疫疗法和 FGFR TKI 在内的联合治疗的潜在益处。最后,鉴于在泛癌队列中观察到的频繁的具有典型特征的 EGFR 融合,研究者提出一项泛癌临床试验,使用EGFR TKI 疗法治疗 EGFR 融合阳性癌症,以帮助确立这种变异作为靶向治疗的生物标志物。这些努力将共同扩大我们对普遍存在于头颈部癌中的可靶向变异的了解,并帮助确立更多用于精准疗法的生物标志物。总之,这些结果构成了对HNC 中基因融合的最大规模综合评估。尽管这些变异较为罕见,但其发生率与临床可靶向的HRAS 变异相当,表明本研究可能有助于为HNC 患者提供更多的治疗机会。
“实体瘤272PLUS+RNA1560基因检测”、“实体瘤DNA1299+RNA1560基因检测”项目,基于DNA+RNA双组学,检测FGFR、EGFR等基因的点突变、插入缺失、融合等多种变异类型,预测可能获益的靶向、免疫、化疗等治疗方案,同时辅助预后和遗传风险评估。
参考文献:
Hoskins EL, Vella R, Reeser JW, et al. Prevalence and biological impact of clinically relevant gene fusions in head and neck cancers. NPJ Precis Oncol. 2025;9(1):221. Published 2025 Jul 3. doi:10.1038/s41698-025-00889-7

